Estudi de la variabilitat dels gens predits
En l'apartat anterior, intentàvem validar els gens predits pel genscan i el geneid amb les bases de dades de ESTs. Però tot i aplicar un filtre sobre els ESTs, la densitat d'aquests és tan gran, que no obtenim una informació prou clara. Per això, per tal d'analitzar amb major precisió les prediccions de cada un dels gens validades amb els spliced ESTs hem decidit de tornar a realitzar tot el procediment anterior amb cada fragment de seqüència en què es prediu un gen.
En primer lloc hem hagut de tallar la seqüència en funció de les coordenades que hem obtingut per cada gen (en els programes de predicció de gens geneid i genscan). Per tal de realitzar aquest procediment hem utilitzat un programa anomenat fastachunk. Aquest programa necessita saber quines són les coordenades de la posició inicial de tall, i quants nucleòtids vols tallar. També cal un pipe amb la comanda fold perquè el tros de seqüència resultant quedi trencat en linies de 60 nucleòtids. Les coordenades que vam utilitzar van ser:
- $ fastachunk seq1.txt 1 36421 | fold -60 > hg16_dna_1-36421
Cal apuntar que en cada cas vàrem agafar 1000 bases per sobre i per sota d'on començava la predicció dels gens.
GEN PREDIT
POSICIÓ INICI
POSICIÓ FINAL
NOM SEQÜÈNCIA
1 genscan/geneid
1
36421
hg16_dna_1-36421
2 genscan/geneid
79118
215671
hg16_dna_79118-215671
3 genscan/geneid
213644
269504
hg16_dna_213644-269504
4 genscan/geneid
261662
338899
hg16_dna_261662-338899
5 i 6 genscan/ 5 geneid
349269
367838
hg16_dna_349269-367838
7, 8 i 9 genscan / 6 geneid
369503
500000
hg16_dna_369503-500000
Un cop vam aconseguir les noves seqüències, ens van disposar a realitzar els mateixos passos que anteriorment estan descrits per la seqüència de 500Kb. En primer lloc però, per treballar amb els fitxers gff del genscan,el geneid,el fgenesh i el grail vam haver de modificar-los amb les noves coordenades. Així a l'hora d'obtenir els gràfics, els gens predits s'aliniarien amb els spliced ESTs. Per fer això vam necessitar l'ordre amb gawk que vam executar sobre els gff de geneid i genscan, per cada gen anteriorment descrit a la taula:
- gawk 'BEGIN {OFS="\t"} $4>= (posició inici del gen ) && $5<= (posició final del gen ) ; {$4=$4 - (posició inici nova seqüència) ; $5=$5 - (posició inici nova seqüència) ; print $0 }' seq1m.genscan.gff o seq1m.geneid.gff o seq1grail.gff o seq1softberry.gff > seq1m.genscan_(número gen).gff o seq1m.geneid_(número gen).gff o seq1grail_(número gen).gff o seq1softberry_(número gen).gff
Un cop tenim tot els fitxers, amb els nous gff per geneid i genscan (un per cada gen), hem d'obtenir els gff per les noves seqüències. Aquest procès fins a l'obtenció dels gràfics, on es mostren cada gen per separat amb les prediccions dels ESTs i geneid i genscan, és anàleg a l'utilitzat anteriorment (pas de gff a gff2ps i visualització amb kview).
Al llarg de la realització del treball hem tingut problemes amb el parseblast. Un cop ja havíem dut a terme tot el procès des de l'obtenció dels ESTs, fins a la visualització de les prediccions amb els ESTs spliced per totes les subseqüències, ens vam adonar que la subseqüència compresa entre els nucleòtids 349269-367838, tenia un error (a nivell de megablast). I al dur a terme la repetició ens va sorgir una complicació amb el parseblast que hem estat incapaces de resoldre. Per tant aquesta subseqüència no ha pogut ser analitzada en aquest apartat.
Per accedir als gràfics:
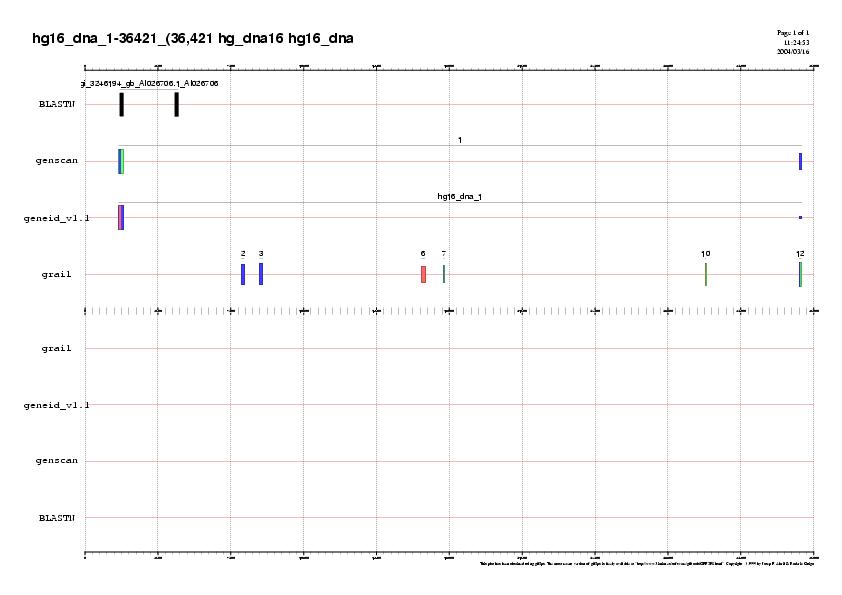
Com es pot observar en els gràfics, no tots els resultats proporcionen nova informació. Així, per exemple, quan representem la primera subseqüència (1-36421), només s'observen dos ESTs un cop s'ha fet el spliced. A part, aquests només ens corroboren el que seria l'extrems 5' del gen predit per geneid i genscan. El fgenesh no predia cap gen en aquesta regió. És per això, que correm un nou megablast amb tots els ESTs (no només els humans). Tanmateix, els resultats no són significativament millors.
- Resultats usant només els ESTs humans:
hg16_dna_1-36421.html
S'observen 21 ESTs, tots ells de transcripts que s'expressen en diferents teixits human.
- Resultats usant tots els ESTs:
hg16_dna_1-36421.t.html
S'observen pràcticament els mateixos ESTs que en el cas anterior, tot i que al blastn li permetiem 1000 descripcions i 1000 aliniaments amb ESTs. Aixó ens fa pensar en quèsi aquest exó que prediuen els programes existeix, possiblement es troba només en humans. Cal pensar també que les bases de dades de ESTs poden no ser completes.
En conclusió, podríem dir que els ESTs no ens validen aquest primer gen que prediuen els programes geneid i genscan.
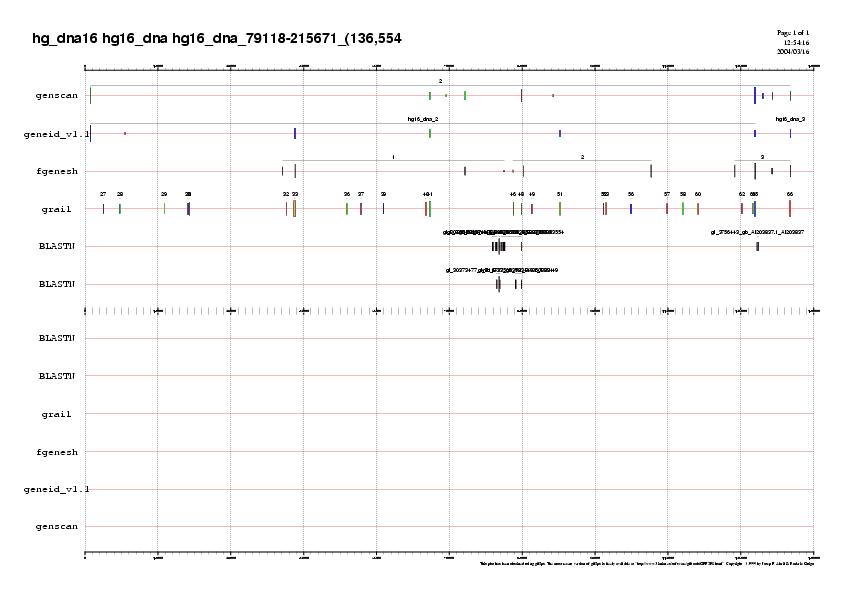
En la segona subseqüència , la representació del genscan, geneid, fgenesh, grail i els ESTs, mostra que només un EST reconeix en l'extrem 3' de les quatre prediccions,un exó. I després hi ha un conjunt de ESTs a una regi&oaute; més central, que en conjunt només permeten corroborar, i de forma no molt clara, alguns dels exons predits. Per intentar millor la informació de què disposem, farem un script per intentar filtrar els ESTs, ja que el blastn pot donar-nos molts ESTs que se solapin pels seus extrems, i aleshores tenim molta informació redundant.
Tanmateix el resultat després d'haver fet el spliced era exactament el mateix al anterior, com es pot comprovar:
hg16_dna_79118-215671.spliced.est+genepredictions.jpg
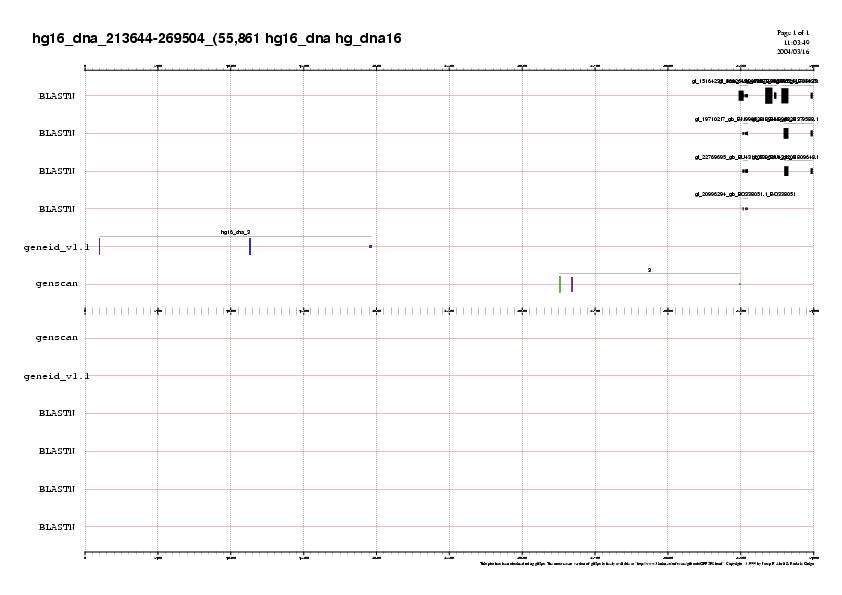
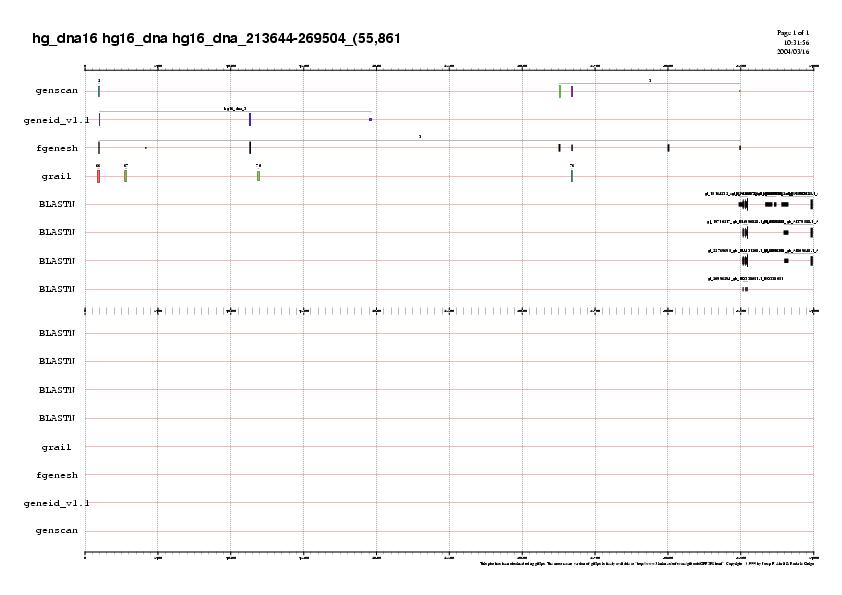
En la subseqüència que conté entre els nucleòtids 213644-269504 , hi ha una regió on se solapen molts dels ESTs. Veiem que aquests ESTs confirmen només un dels tres exons (l'últim) que el genscan prediu en el gen 3. En canvi, no ens prediuen cap dels exons del geneid. El que farem és, d'una banda, afegir la informació que ens proporcionaven el grail i el fgenesh hg16_dna_213644-269504.tots.gff, i de l'altra, aplicarem el mateix programa que en el cas anterior per filtrar i repuntuar els ESTs.
Per veure el gràfic :
hg16_dna_213644-269504.genepredicitions.definitiu.jpg
En aquest cas, el que veiem es que no se'ns filtren els ESTs perquè no deuen començar a les mateixes coordenades. Peró a més veiem que els ESTs també confirmen el sisè exó predit per fgenesh.
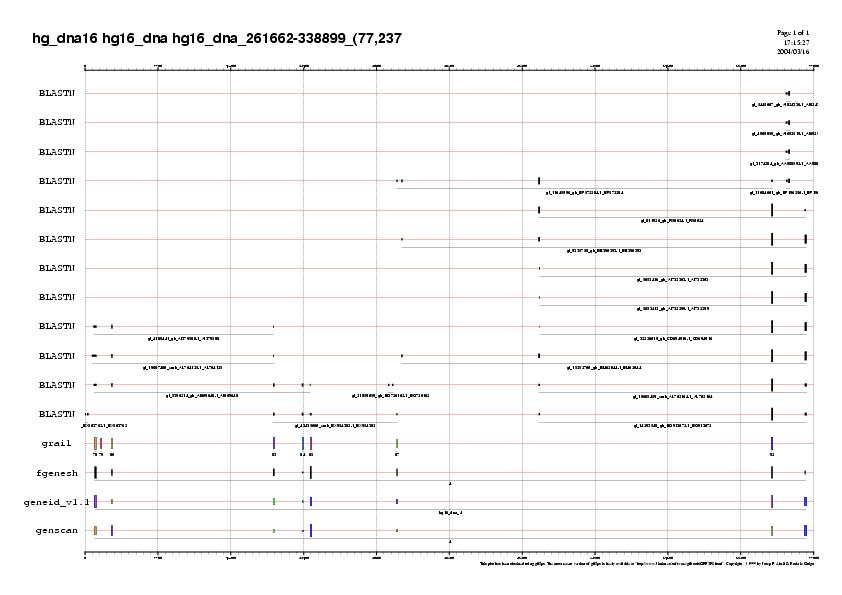
Una de les altres subseqüències amb que hem treballat és la que està formada pels nucleòtids 261662-338899 . Al gràfic anterior s'observa que hi ha molts ESTs que ens soporten els exons predits en reverse pels diferents programes. Per exemple, estan soportats:
- els exons 2, 4, 5, 6, 7, 8, 9 i 10 del grail
- els exons 1, 2, 3, 4, 5, 6, 7, 8 i 10 de fgenesh.
- els exons 1, 2, 4, 5, 6, 7, 8 i 10 geneid i del genscan.
Però ens estranya molt que havent-hi tants ESTs que prediuen un exó entre els exons 2 i 3 de geneid i genscan, els programes no en prediuen cap.
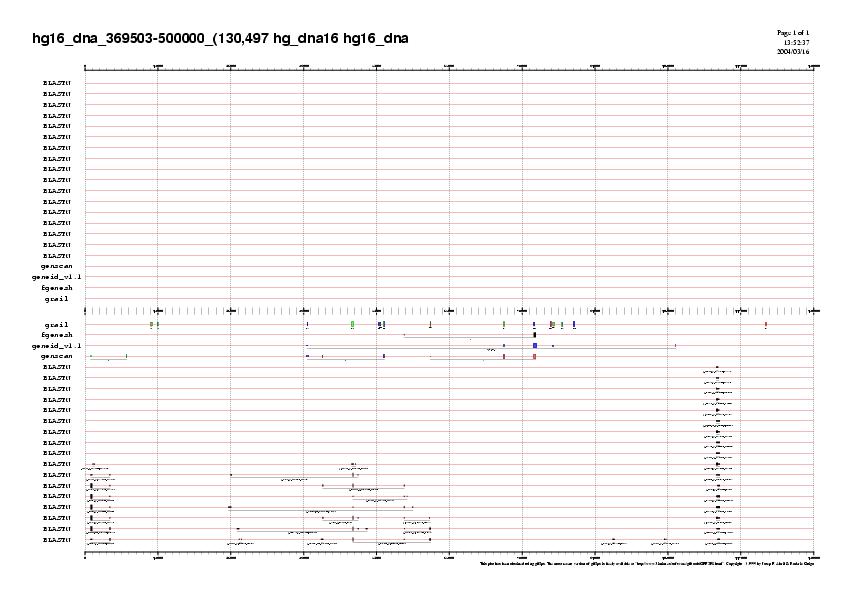
En el darrer gràfic ,en que consideràvem el gen 6 de geneid, i els gens 7,8 i 9 de genscan, que estan en reverse, apareixen molts ESTs i ens interessa buscar més informació. Per això farem el mateix script per intentar filtrar els ESTs (blastn pot donar-nos molts ESTs que se solapin pels seus extrems, i aleshores tenim molta informació redundant)
I veiem exactament el mateix que abans de filtrar els ESTs:
hg16_dna_369503-500000.spliced.genepredictions.jpg>
Això pot ser degut al fet que els ESTs no tinguin les mateixes coordenades d'inici i/o final, i aleshores no quedarien filtrats amb el script. En les pàgines següents, realitzarem una anàlisi més detallat de la regió central del gràfic, on hi ha aquest conjunt de ESTs que només prediuen l'exó 2 del gen 8 de genscan.
A partir dels ESTs, ens adonem que en aquesta regió hi ha fenomens de splicing alternatiu, perquè, determinats ESTs, prediuen una combinació d'exons diferents entre els diferents ESTs, i també respecte als programes.

Cal apuntar que en cada cas vàrem agafar 1000 bases per sobre i per sota d'on començava la predicció dels gens.
| GEN PREDIT | POSICIÓ INICI | POSICIÓ FINAL | NOM SEQÜÈNCIA |
| 1 genscan/geneid | 1 | 36421 | hg16_dna_1-36421 |
| 2 genscan/geneid | 79118 | 215671 | hg16_dna_79118-215671 |
| 3 genscan/geneid | 213644 | 269504 | hg16_dna_213644-269504 |
| 4 genscan/geneid | 261662 | 338899 | hg16_dna_261662-338899 |
| 5 i 6 genscan/ 5 geneid | 349269 | 367838 | hg16_dna_349269-367838 |
| 7, 8 i 9 genscan / 6 geneid | 369503 | 500000 | hg16_dna_369503-500000 |
Un cop vam aconseguir les noves seqüències, ens van disposar a realitzar els mateixos passos que anteriorment estan descrits per la seqüència de 500Kb. En primer lloc però, per treballar amb els fitxers gff del genscan,el geneid,el fgenesh i el grail vam haver de modificar-los amb les noves coordenades. Així a l'hora d'obtenir els gràfics, els gens predits s'aliniarien amb els spliced ESTs. Per fer això vam necessitar l'ordre amb gawk que vam executar sobre els gff de geneid i genscan, per cada gen anteriorment descrit a la taula:
- gawk 'BEGIN {OFS="\t"} $4>= (posició inici del gen ) && $5<= (posició final del gen ) ; {$4=$4 - (posició inici nova seqüència) ; $5=$5 - (posició inici nova seqüència) ; print $0 }' seq1m.genscan.gff o seq1m.geneid.gff o seq1grail.gff o seq1softberry.gff > seq1m.genscan_(número gen).gff o seq1m.geneid_(número gen).gff o seq1grail_(número gen).gff o seq1softberry_(número gen).gff
Un cop tenim tot els fitxers, amb els nous gff per geneid i genscan (un per cada gen), hem d'obtenir els gff per les noves seqüències. Aquest procès fins a l'obtenció dels gràfics, on es mostren cada gen per separat amb les prediccions dels ESTs i geneid i genscan, és anàleg a l'utilitzat anteriorment (pas de gff a gff2ps i visualització amb kview).
Al llarg de la realització del treball hem tingut problemes amb el parseblast. Un cop ja havíem dut a terme tot el procès des de l'obtenció dels ESTs, fins a la visualització de les prediccions amb els ESTs spliced per totes les subseqüències, ens vam adonar que la subseqüència compresa entre els nucleòtids 349269-367838, tenia un error (a nivell de megablast). I al dur a terme la repetició ens va sorgir una complicació amb el parseblast que hem estat incapaces de resoldre. Per tant aquesta subseqüència no ha pogut ser analitzada en aquest apartat.
Per accedir als gràfics:
Com es pot observar en els gràfics, no tots els resultats proporcionen nova informació. Així, per exemple, quan representem la primera subseqüència (1-36421), només s'observen dos ESTs un cop s'ha fet el spliced. A part, aquests només ens corroboren el que seria l'extrems 5' del gen predit per geneid i genscan. El fgenesh no predia cap gen en aquesta regió. És per això, que correm un nou megablast amb tots els ESTs (no només els humans). Tanmateix, els resultats no són significativament millors.
- Resultats usant només els ESTs humans:
hg16_dna_1-36421.html
S'observen 21 ESTs, tots ells de transcripts que s'expressen en diferents teixits human.
- Resultats usant tots els ESTs:
hg16_dna_1-36421.t.html
S'observen pràcticament els mateixos ESTs que en el cas anterior, tot i que al blastn li permetiem 1000 descripcions i 1000 aliniaments amb ESTs. Aixó ens fa pensar en quèsi aquest exó que prediuen els programes existeix, possiblement es troba només en humans. Cal pensar també que les bases de dades de ESTs poden no ser completes.
En conclusió, podríem dir que els ESTs no ens validen aquest primer gen que prediuen els programes geneid i genscan.
En la segona subseqüència , la representació del genscan, geneid, fgenesh, grail i els ESTs, mostra que només un EST reconeix en l'extrem 3' de les quatre prediccions,un exó. I després hi ha un conjunt de ESTs a una regi&oaute; més central, que en conjunt només permeten corroborar, i de forma no molt clara, alguns dels exons predits. Per intentar millor la informació de què disposem, farem un script per intentar filtrar els ESTs, ja que el blastn pot donar-nos molts ESTs que se solapin pels seus extrems, i aleshores tenim molta informació redundant.
Tanmateix el resultat després d'haver fet el spliced era exactament el mateix al anterior, com es pot comprovar:
hg16_dna_79118-215671.spliced.est+genepredictions.jpg
En la subseqüència que conté entre els nucleòtids 213644-269504 , hi ha una regió on se solapen molts dels ESTs. Veiem que aquests ESTs confirmen només un dels tres exons (l'últim) que el genscan prediu en el gen 3. En canvi, no ens prediuen cap dels exons del geneid. El que farem és, d'una banda, afegir la informació que ens proporcionaven el grail i el fgenesh hg16_dna_213644-269504.tots.gff, i de l'altra, aplicarem el mateix programa que en el cas anterior per filtrar i repuntuar els ESTs.
Per veure el gràfic :
hg16_dna_213644-269504.genepredicitions.definitiu.jpg
{kind=link}
En aquest cas, el que veiem es que no se'ns filtren els ESTs perquè no deuen començar a les mateixes coordenades. Peró a més veiem que els ESTs també confirmen el sisè exó predit per fgenesh.
Una de les altres subseqüències amb que hem treballat és la que està formada pels nucleòtids 261662-338899 . Al gràfic anterior s'observa que hi ha molts ESTs que ens soporten els exons predits en reverse pels diferents programes. Per exemple, estan soportats:
- els exons 2, 4, 5, 6, 7, 8, 9 i 10 del grail
- els exons 1, 2, 3, 4, 5, 6, 7, 8 i 10 de fgenesh.
- els exons 1, 2, 4, 5, 6, 7, 8 i 10 geneid i del genscan.
Però ens estranya molt que havent-hi tants ESTs que prediuen un exó entre els exons 2 i 3 de geneid i genscan, els programes no en prediuen cap.
En el darrer gràfic ,en que consideràvem el gen 6 de geneid, i els gens 7,8 i 9 de genscan, que estan en reverse, apareixen molts ESTs i ens interessa buscar més informació. Per això farem el mateix script per intentar filtrar els ESTs (blastn pot donar-nos molts ESTs que se solapin pels seus extrems, i aleshores tenim molta informació redundant)
I veiem exactament el mateix que abans de filtrar els ESTs:
hg16_dna_369503-500000.spliced.genepredictions.jpg>
Això pot ser degut al fet que els ESTs no tinguin les mateixes coordenades d'inici i/o final, i aleshores no quedarien filtrats amb el script. En les pàgines següents, realitzarem una anàlisi més detallat de la regió central del gràfic, on hi ha aquest conjunt de ESTs que només prediuen l'exó 2 del gen 8 de genscan.
A partir dels ESTs, ens adonem que en aquesta regió hi ha fenomens de splicing alternatiu, perquè, determinats ESTs, prediuen una combinació d'exons diferents entre els diferents ESTs, i també respecte als programes.