

Ana Igea Fernández, Iris Uribesalgo Micás
Los datos de los que partimos eran 60770 cDNAs de ratón, dentro de los cuales había algunos descritos como codificantes, otros como no codificantes y teóricamente todos estaban clonados en forward (link al artículo de referencia).
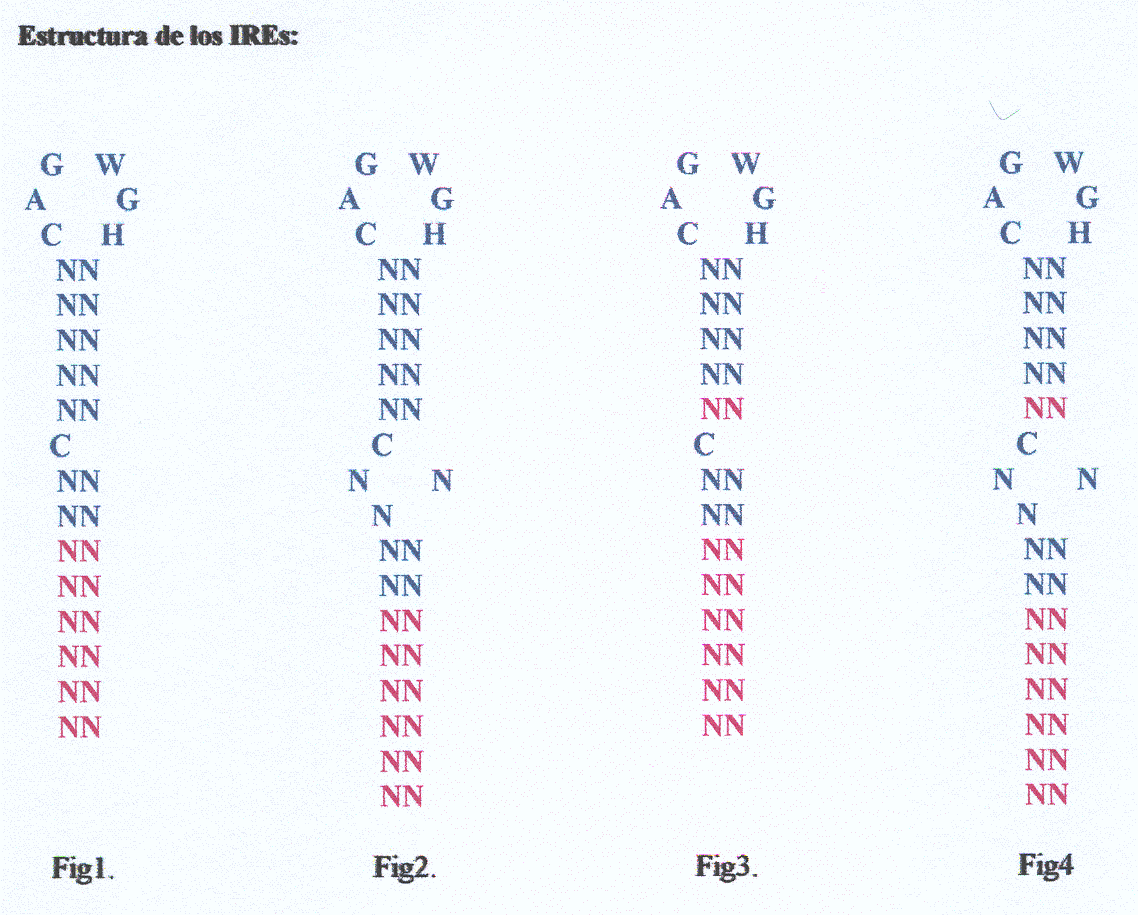
Para comenzar nuestro trabajo lo primero que necesitamos fue crear un patrón que nos definiera las estructuras de los IREs que estábamos interesados en encontrar. A partir de los patrones de IREs realizados el año anterior y basándonos también en la bibliografía que encontramos en diferentes artículos [1,2,3,4,5,6,7,8], decidimos utilizar dos patrones de predicción diferentes:
Decidimos utilizar en un primer momento dos patrones ya que, dependiendo del tipo de proteína, los patrones de IREs difieren considerablemente (ver patrones), de manera que pensamos que, quizás, con un patrón más laxo que el primero podríamos incluir los IREs de una gran mayoría de proteínas sino de todas. Para acabar de definir la estructura de este patrón nos basamos en los siguientes datos encontrados en la literatura: Al utilizar dos patrones nos planteamos la posibilidad de que el nuevo patrón más laxo hiciera aumentar el número de resultados obtenidos y, por lo tanto, nos hiciera tener un mayor número de falsos positivos. Deberíamos valorar al obtener los primeros resultados de posibles estructuras IRE si este patrón laxo nos era de utilidad o, si por el contrario, nos incluía demasiadas estructuras no específicas.
Utilizamos el programa Scan_for_matches para correr los patrones sobre los cDNAs de partida.
La primera disyuntiva que se nos planteó en este punto fue si debíamos correr el programa sobre los 60770 cDNAs de ratón o solamente sobre los cDNAs descritos como codificantes y, además, si debíamos correrlo únicamente en forward o también en reverse.
En primer lugar decidimos utilizar los 60770 cDNAS de ratón para estar seguros de que no encontramos ningún IRE en regiones no codificantes y de que realmente las regiones que el artículo de referencia cualifica de no codificantes lo son. En segundo lugar, decidimos correr en forward así como también en reverse cada uno de los patrones y poner los resultados en archivos diferentes para su posterior análisis (IRESpatroreverse.txt, IRESpatroforward.txt, IRESpatrolaxereverse.txt, IRESpatrolaxeforward.txt):
Ante estos resultados decidimos seguir trabajando con las secuencias obtenidas a partir del patrón laxo puesto que incluye un mayor número posible de estructuras IREs y la diferencia respecto a las secuencias obtenidas con el patrón estricto tampoco es tan elevada (601 IREs en forward vs. 191 IREs en forward). Lo único que debemos tener en cuenta en este caso es que tendremos que ser bastante estrictos cuando se haga la validación termodinámica (link al trabajo), y cuando hagamos la validación por homología con humano (Blast).
La siguiente decisión que tomamos fue trabajar, dentro de las secuencias obtenidas a partir del patrón laxo, solamente con los resultados en forward. Dicha decisión se tomó apoyada por los resultados del grupo encargado de la predicción de genes (link al trabajo), el cual ejecutó el programa "geneid" con los 60770 cDNAs deduciendo que, aunque había algunos cDNAs en reverse, estos eran pocos y la mayoría no tenían buenas puntuaciones. Basándonos en estos datos, y aunque algún cDNA en reverse tenía una puntuación buena, creímos que era mejor trabajar solamente en forward aunque pudiéramos perdernos alguna información por la complicación añadida que contrae trabajar en reverse frente a la pequeña posibilidad de encontrar alguna estructura IRE en dichos cDNAs.
Al tener aclarados y decididos todos los puntos anteriores, trabajamos con el fichero de las secuencias obtenidas en forward a partir del patrón laxo que contiene una predicción de 601 IREs. En este momento fue necesario transformar el fichero que teníamos en lenguaje Patscan a lenguaje Fasta para que pudiera ser validado con otros programas que requieren dicho lenguaje. Para ello, creamos un programa en PERL denominado RNAfold_fasta.pl, el cual convertía nuestras secuencias eliminando los espacios entre nucleótidos y cambiándonos las Ts por Us (IRESpatrolaxeforward.fasta).
Para validar nuestros resultados realizamos un Blast de las 601 secuencias correspondientes a posibles IREs contra el genoma humano puesto que la mayoría de los IREs encontrados en ratón deberían hallarse también en humano (el 99% del genoma esta conservado y los IREs, además, sobreviven al splicing y tienen una función concreta).
"Blast" es un recurso que podemos encontrar en la web o que podemos correr directamente des de UNIX. Nosotros utilizamos la aplicación desde UNIX además de crear dos variables [4] (una para el patrón y otra para la base de datos) que deberíamos introducir cada vez al entrar al shell , puesto que no las guardaba en memória, para ahorrarnos hacer un bucle con la finalidad de que el recurso Blast recorriera todos los cromosomas.
Antes de utilizar Blast debíamos decidir:
Después de razonar y definir todo los parámetros nos dispusimos a correr el programa Blast de las 601 secuencias de posibles IREs sobre la base de datos del genoma humano enmascarado pero no fue posible acabarlo puesto que el ordenador con el que trabajábamos (persy) no soportó esta carga y falló. Frente a este echo decidimos correr el Blast en otro ordenador especial para utilizar dicho programa llamado blastmachine, el cual es más potente y esperábamos que pudiera soportar el tamaño de la base de datos [5].
Lo que parecía una solución tampoco resultó eficaz y el programa siguió sin darnos resultados puesto que la blastmachine también nos dió problemas, de manera que decidimos intentar hacer el Blast cromosoma por cromosoma manteniendo el valor de 0.1 de Expected value [6], pero los resultados siguieron siendo negativos puesto que los ordenadores no soportan una base de datos de un tamaño tan grande.
El último recurso fue intentar reducir el tamaño de la base de datos utilizando ESTs humanos en lugar de todo el genoma entero enmascarado. Dicho recurso tiene sentido puesto que los IREs se transcriben y sobreviven al proceso de splicing a la vez que eliminamos intrones y regiones intergénicas que pueden dar un gran número de falsos positivos. Esta vez sí fue posible realizar el Blast pero los resultados obtenidos, debido aún al gran tamańo de la base de datos, fueron poco significativos (resultados).
En este momento nos planteamos la posibilidad de correr nuestro patrón laxo sobre una base de datos de cDNAs humanos creada a partir de ESTs humanos disponible en la web para posteriormente utilizar las secuencias que obtuviéramos como base de datos para hacer un nuevo Blast con las 601 secuencias obtenidas con este mismo patrón a partir de los 60770 cDNAs de ratón [7]. De esta manera conseguimos, por una parte, trabajar solo con regiones codificantes (evitando intrones y regiones intergénicas) y, por la otra, reducir el tamaño de la base de datos esperando obtener hits más específicos y menos falsos positivos. Sin embargo, también fue imposible realizar este Blast, ya que existe algún problema ya sea debido al tamaño de la base de datos (654010 secuencias y 340650979 nucleótidos) o a un problema interno del propio ordenador. Por ello, el único resultado que pudimos obtener fue el mostrado anteriormente usando la base de datos de ESTs el cual ya hemos dicho que no es demasiado significativo (tenemos valores de E-value aún muy elevados).
Conseguimos crear un patrón a partir del cual obtuvimos una serie de secuencias que contenían genes con estructuras secuandarias en el 3' o el 5' UTR que podrían corresponder a IREs reales. Esta predicción se realizó sobre los 60770 cDNAs de ratón y los resultados se validaron por homología mediante un BLAST contra genoma humano que al final se tuvo que llevar a cabo contra una base de datos de ESTs humanos. Esta validación por BLAST tiene sentido ya que se ha comprovado mediante comparación de secuencias animales que contenían IREs que la conservación de identidad de secuencia es mucho mayor (más del 90%) entre especies para el mismo mRNA que entre diferentes mRNAs dentro de la misma especie (36-85% identidad).
Los resultados obtenidos después de todo este proceso no fueron demasiado significtivos como ya hemos indicado aunque ha sido por causas externas a nosotras. De todas maneras, debemos tener en cuenta que esta sólo es una pequeña parte del conjunto del trabajo los resultados finales del cuál se muestran en el siguiente link.
2-03-2003 E-mail: anuky4@hotmai.com ; yurcelay@hotmail.com
Con este patrón descrito en la literatura [1], como muestra la figura, esperamos encontrar cinco apareamientos exactos en la "stem" superior y de dos a ocho apareamientos en la "stem" inferior. Además, aceptamos los apareamientos A-T y G-C así como los G-T ya que se ha observado que, aunque estos últimos no sean apareamientos clásicos, se encuentran en un gran número de estructuras secundarias.
Este patrón, que definimos nosotros a partir de los datos encontrados en la literatura, difiere del anterior en que se permiten cuatro o cinco apareamientos en la "stem" superior.
2. Ejecución del programa "Scan_for_matches"
3. Traducción de los resultados a lenguaje FASTA
4. BLAST contra genoma humano
5. Resultados
