

Ana Igea Fernández, Iris Uribesalgo Micás
We started from 60.770 mouse cDNAs (some of them were described as codificants, other as non codificants and theorically all of them were cloned in forward) (link to the reference article).
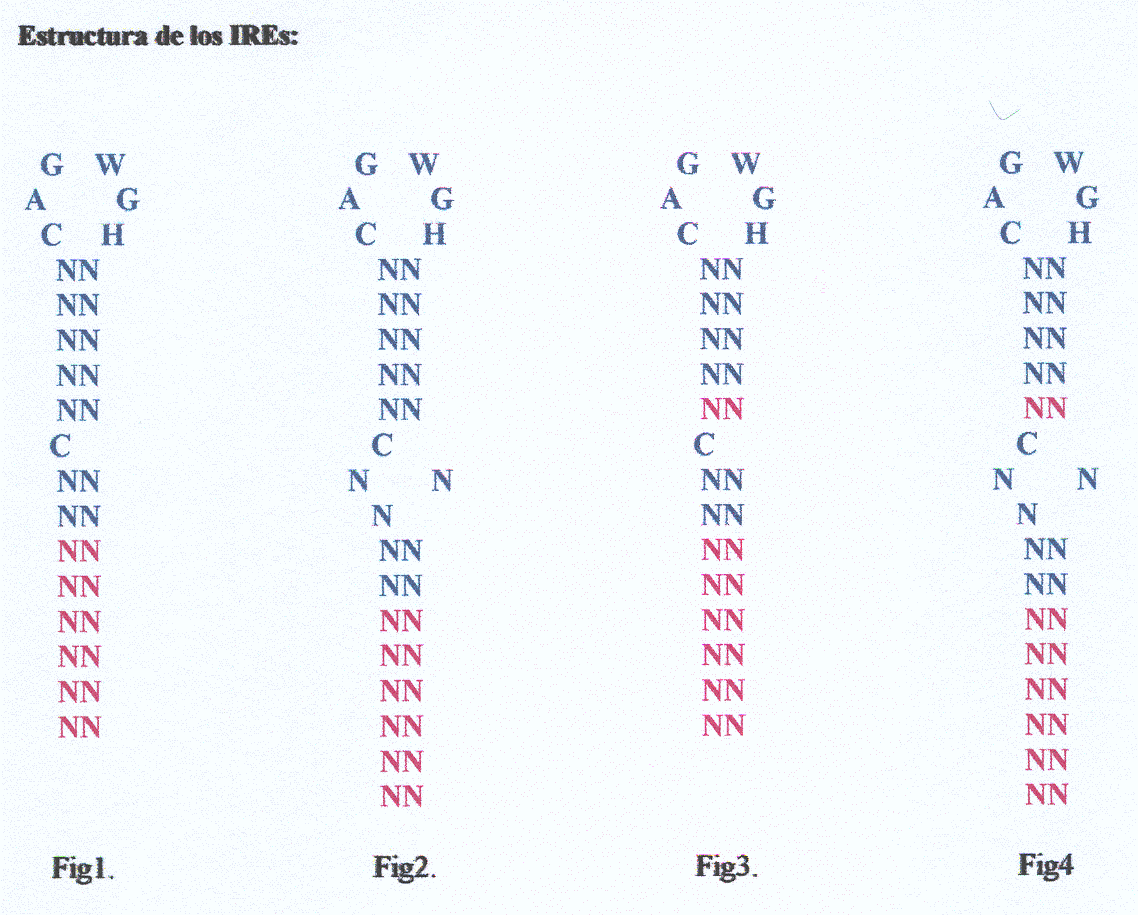
First of all we created a motif that defined those IREs structures we wanted to find. Starting from the IREs motif created last year and taking in account scientific bibliography finded in different articles [1,2,3,4,5,6,7,8], we decided to use two diferent motifs to make our prediction:
This motif described in our bibliography [1] is useful to find five exact matches in the upper stem and from two to eight matches in the lower stem (as the figure show). Moreover, we accept A-T and G-C matches as well as G-T matches because it has been observed that although the last matches are not the classical ones are also present in a big number of secondary structures.
This standard was defined taking in account some data found in scientific literature. It is different from the other one and allows four or five matches in the upper stem.
At the beginning we decided to use both motifs to compare their results because it's well known that in each protein motifs change (see motifs variation). Our hypothesis was that using the relaxed pattern we could include a lot of proteins containing IREs. To define completely the structure of the relaxed motif we searched some bibliography and took some conclusions:
As we have said before, we used both motifs so it could be possible that because of using the relaxed pattern our results were containing too much false positives. Because of that was important to think if we should use this relaxed motif or just the strict one when obtaining both results.
We used the program "Scan_for_matches" to run our patterns in the initial cDNAs.
First of all we decided if we should use all mouse cDNAs (60770 sequences) or only those described as codificants and if we should run the program in forward or also in reverse.
Our first decision was using all the mouse cDNAs to be sure about it wasn't any IRE in non-codificant regions and also to certify that was true that this regions described in the reference article were non-codificant. Secondly we chose run both patterns in forward and in reverse having the results in separately files to analyze them later (IRESpatroreverse.txt, IRESpatroforward.txt, IRESpatrolaxereverse.txt, IRESpatrolaxeforward.txt):
When having these results we decided going on with the sequences obtained from the relaxed motif because it includes a bigger number of possible IREs structures and also because the difference between the results obtained with this pattern and the ones obtained with the strict one were not too important (601 IREs in forward vs. 191 IREs in forward). Because of that it was important to us keeping in mind that we should be strict enough when doing the termodinamic and the homology using human DNA (Blast) validations.
Our next dicision was working only with those sequences obtained by running the program in forward using the relaxed pattern. This decision was taken because of the gene prediction results that were obtained by another group running the program called "geneid". They concluded that the main part of mouse cDNAs were cloned in forward although also found some cloned in reverse (but in general had a low score). This information helped us to decide that was better working only in forward (we could lose information but it would be easier and the possibility of finding an IRE structure in reverse were very low).
When having the previous points decided and clear, we used the file containing the sequences obtained in forward by the relaxed motif (601 IREs predicted and comming from 594 different cDNAs). After that was necessary to transform the file in Patscan languaje to Fasta languaje to be validated with other programs that requiered this kind of languaje. In order to make this transformation we created a PERL program called RNAfold_fasta.pl which consisted in transforming our sequences removing spaces between nulceotides and changing Ts by Us (IRESpatrolaxeforward.fasta).
To validate our results we used Blast to compare our 601 predicted IREs sequences with human genome because the main part of IREs finded in mouse should be also finded in human genome (99% of human and mouse genomes were conserved and, moreover, IREs outlast to splicing and have an specific function).
"Blast" is a program we could find in the web or running it directly from UNIX. We used LINUX application and we created two modifiers [4] (one for the pattern and the other for the database) that we should enter every time we opened shell because it doesn't keep them in memory (we used them to avoid a loop and to run Blast over all cromosomes).
Before using Blast we had to decide:
After arguing and defining all the parameters we began to run Blast over the 601 sequences of possible IREs and human masked genome but it wasn't possible because the computer that we used (called persy) didn't carry it and failed. We had the possibilty of running Blast in another computer specially apropiate to do it called blastmachine (it is more potent than persy and we thought it could carry the database) [5]. However, it wasn't a solution and we didn't obtained any result because blastmachine also had problems.
After that we run Blast cromosome by cromosome keeping 0.1 as Expected value [6] but results were also negative because computers didn't support such a big database.
Our last option was trying to reduce the database size using human ESTs instead of human masked genome. We chose this option because IREs are transcribed and outlast to splicing and at the same time using this database we remove introns and intergenic regions which could create false positive matches. This time running Blast was possible but the results we obtained (due to the big size of the database) were not very significant (see results with a not enough low E-value).
To get more significant results we thought about the possibility of running our relaxed pattern over a cDNA database created from human ESTs (it is available in the web) and using this results as a database to run another Blast with our 601 sequences [7]. On one hand, trying this option we work only with codifing regions (avoiding introns and intergenic regions) and, on the other hand, we reduce the size of the database expecting more specific hits and less false positives.
However, was impossible obtaining results because there was some problem. Maybe is an internal computer error or maybe the database is too big (654010 sequences and 340650979 nucleotids). Because of that our uniq result was the one described before (using the ESTs database) although it's not very significant (E-vaules are too high).
We created a motif and we obtained some sequences containing genes with secondary structures in 3' or 5' UTRs that could match with real IREs. This prediction was made over 60770 mouse cDNAs and the results were validated by homology (Blast to human genome) using at the end ESTs human sequences as database. This Blast validation is coherent because comparisons of animal IRE sequences reveal that the conservation of sequence identity is much higher (90% identity) between species for the same mRNA than between different mRNAs in the same species (36-85% identity). The results obtained after all the process weren't as we have said too significant although it was because of external factors. However, we should remind that this is just a small part of the entire project so you can see the final results in this link.
2-03-2003 E-mail: anuky4@hotmai.com ; yurcelay@hotmail.com
2.Run "Scan_for_matches"
3. Results traduction to FASTA languaje
4. BLAST to human genome
5. Results
