

Ana Igea Fernández, Iris Uribesalgo Micás
I dati da cui abbiamo cominciato erano 60.770 cCDNAs di ratto, dentro dei quali ce n'erano alcuni descritti come codifianti, altri como non codificanti e teoricamente tutti erano clonati in forward (articoli).
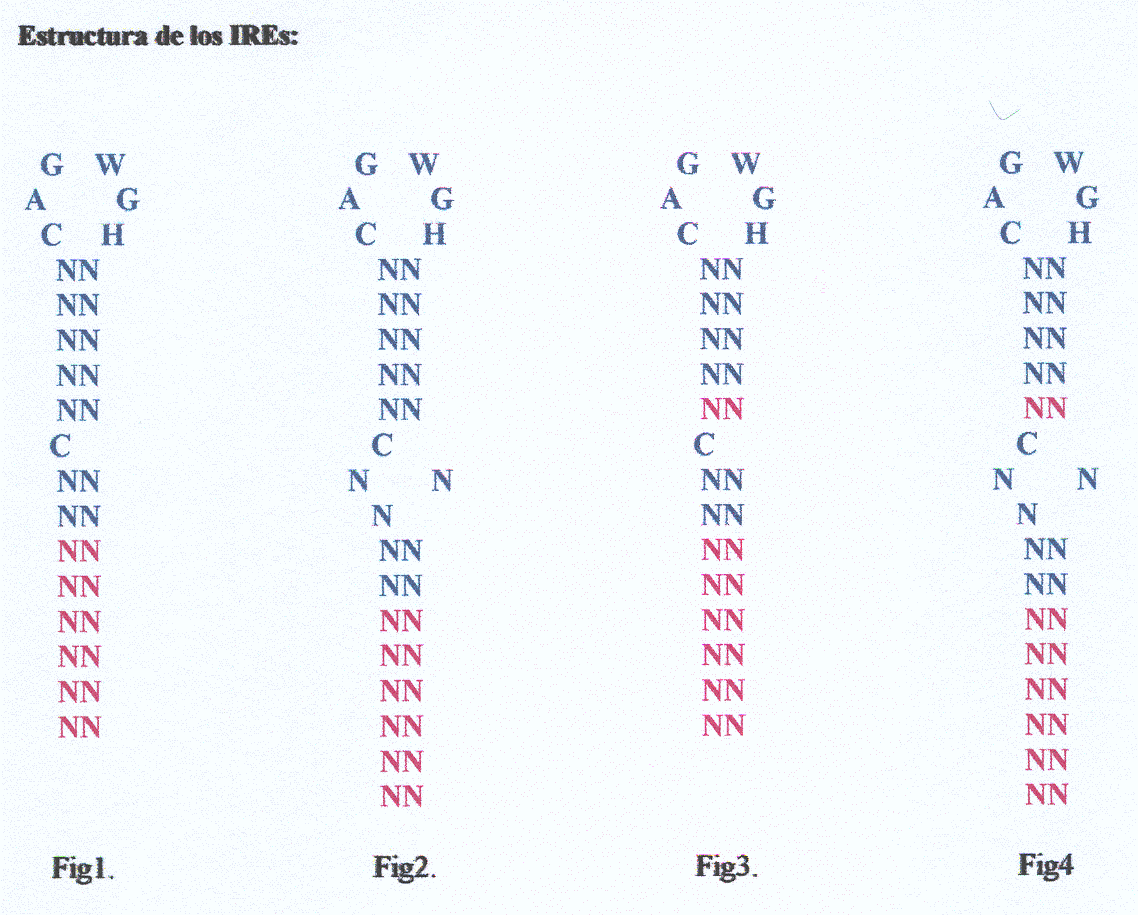
Per cominciare il nostro lavoro la prima cosa di cui abbiamo avuto bisogno è stata creare un modello che ci potesse definire le strutture degli IRE che eravamo interessati a trovare. Utilizzando come base i modelli di IRE realizzati l'anno anteriore ed anche la bibliografi che abbiamo trovato in vari articoli [1,2,3,4,5,6,7,8], abbiamo deciso di utilizzare due modelli di previsioni diversi:
Abbiamo deciso di utilizzare in un primo momento due modelli perchè, dipendendo del tipo di proteina, i modelli di IRE sono considerevolmente diversi (diversi modelli). È per questo che abbiamo pensato che, forse, con un modello più rilassati dal primo si potevano includere gli IRE di una grande maggioranza di proteine, e forse anche di tutte. Per finire di definire la struttura di questo modello abbiamo preso come base questi dati trovati nella bibliografia:
Usando due modelli ci possiamo chiedere la possibilità che il nuovo modello più rilassato possa fare crescere il numero di risultati ottenuti e, quindi, possa farci avere un numero maggiore di risultati positivi. Quindi, dobbiamo valorare, quando si otteneranno i primi risultati de possibili structure IRE se questo modello rilassato ci era utile o se ci includeva troppe strutture non specifiche.
Abbiamo utilizzato il programma "Scan_for_matches" per esecutare il modello su i cDNA d'inizio.
Il primo dilemma che abbiamo trovato in questo punto era se si doveva esecutare il programma su i 60.770 cDNA di ratto o soltanto su i cDNA descritti come codificanti e, in più, se si doveva esecutare unicament in forward, oppure anche in reverse.
Per cominciare, abbiamo deciso di utilizzare i 60.770 cDNA di ratto per essere sicuri che non abbiamo trovato nessun IRE in regioni non codificanti, e che veramente la regioni che l'articolo di riferimento qualifica come non codificanti lo sono. Poi, abbiamo deciso di esecutare il programma in forward ed anche in reverse per ognuno dei modelli, e salvare i risultati in archivi diversi per uno studio posteriore (IRESpatroreverse.txt, IRESpatroforward.txt, IRESpatrolaxereverse.txt, IRESpatrolaxeforward.txt):
Davanti a questi risultati, abbiamo deciso di continuare lavorando con le sequenze ottenute dal modello rilassato, perchč include in maggiore numero possibile di strutture IRE, e perchè la differenza con le sequenze ottenute col modello rigoroso non č che dovremo essere abbastanza rigorosi quando si faccia la valorazione termodinamica, e quando si faccia
la valorazione per omologia con umano (Blast).
La prossima decisione che abbiamo preso č stata lavorare, dentro delle sequenze ottenute dal patrone rilassato, soltanto con i risultati in forward. Questa decisone č stata presa appoggiata per i risultati del gruppo che ha lavorato sulla previsione di gen (link), che ha esecutato il programma "geneid" coi 60.770 cDNA facendo la deduzione che, anche se c'erano alcuni cCDNA in reverse, questi cDNA erano pochi e la maggioranza non avevano buoni punteggi.
Da questi dati, e anche se c'era qualche cDNA in reverse che aveva un buon punteggio, abbiamo pensato che era meglio lavorare soltando in forward anche se si poteva perdere qualche informazione per la difficoltà di lavorare in reverse di fronte alla piccola possibilità di trovare qualche struttura IRE nei cDNA.
Una volta chiarti e decisi tutti i punti anteriori, abbiamo lavorato con l'archivo delle sequenze ottenute in forward dal patrone rilassato, che contiene una previsione di 601 IRE, che provengono di 594 cDNA diversi. In questo momento abbiamo avuto bisogno di transformare l'archivio che avevamo in linguaggio fold a linguaggio fasta, per potere essere validato con altri programmi che chiedono questo linguaggio. Per questo abbiamo creato un programma in PERL, che abbiamo nominato RNAfold_fasta.pl che convertiva le nostre sequenze eliminando gli apai fra nucleotidi e cambiando le T per U (IRESpatrolaxeforward.fasta).
Per validare i nostri risultati abbiamo realizzato un Blast delle 601 sequenze corrispondenti a possibili IRE contro il genoma umano, dato che la maggioranza degli IRE trovati in ratto si dovrebbero trovare anche in umano (il 99% del genoma č conservato e gli IRE, in piů, sopravvivono allo splicing ed hanno una funzione concreta).
"Blast" č una risorsa che si piů trovare nella web oppure esecutabile direttamente da UNIX. Noi abbiamo deciso di utilizzare questa risorsa da UNIX, e per questo abbiamo utilizzato un commando specifico [4], ed in piů abbiamo creato una variable per evitare di creare un bucle. Cosě la risorsa Blast puň atraversare tutti i cromosomi.
Prima di utilizzare Blast dovevamo decidere:
Dopo ragionare e definire tutti i parametri abbiamo esecutato il programma Blast delle 601 sequenze di possibili IRE sulla database del genoma umano mascherato, ma non č stato possibile finirlo perchè il computer con cui lavoravamo (persy) non ha potuto sostenere questa carica e si è bloccato. Davanti a questo fatto, abbiamo deciso di esecutare il Blast in un altro computer speciale per utilizarre questo programma il "Blastmachine", che č più potente. Speravamo che il computer potrebbe sostenere il formato della database [5].
Questo sembrava una soluzione ma non è stata neanche efficace e il programma ha continuato a non darci risultati visto che la Blastmachine ci ha dato anche dei problemi. Così, abbiamo deciso di intentar fare il Blast cromosoma per cromosoma, conservando il valore di 0.1 di Expected Value [6], ma i risultati hanno continuato ad essere negativi visto che i computer non sostengono una database di un formato così grande.
L'ultima risorsa è stata cercare di ridurre il formato della database utilizzando EST umani, invece di tutto il genoma intero mascherato. Questa risorsa a un senso, perchè gli IRE si trascrivono e sopravvivono al processo di splicing, ed allo stesso tempo eliminiamo introni e regioni intergeniche che ci possono dare un numero importante di falsi positivi.Questa volta sí che è stato possibile realizzare il Blast ma i risultati ottenuti, visto ancora il grande formato della database, non sono stati molto significativi (risultati).
In questo momento abbiamo pensato la possibilitŕ di esecutare il nostro modello rilassato su una database di cDNA umani creata da EST umani, disponibile sulla web, per poi utilizzare le sequenze che abbiamo ottenuto come database per fare un nuovo Blast con le 601 sequenze ottenute con questo stesso modello dai 60.770 cDNA di ratto [7]. Cosě, abbiamo potuto, di un lato, lavorare soltanto con regioni codificanti (evitando introni e regioni intergeniche), e dell'altro, ridurre il formato della database sperando di ottenere hits piů specifichi e meno falsi positivi. Ma č stato impossibile, anche, realizzare questo Blast, perche esiste qualche problema, forse per il formato della database (654.010 sequenze e 340.650.979 nucleotidi) o forse per un problema interno della stessa computer. Per questo, l'unico risultato che abbiamo potuto ottenere č stato quello che abbiamo mostrato prima, utilizzando la database di EST, che abbiamo detto giŕ che non č molto significativo (abbiamo valori di E-value ancora molto elevati).
Abbiamo potuto creare un modello da cui abbiamo ottenuto diverse sequenze che contenevano gen con strutture secondarie nel 3' o nel 5' UTR, che potrebbero coincidere con IRE reali. Questa previsione si è realizzata sui 60.770 cDNA di ratto ed i risultati sono validati per omologia con un BLAST contro genoma umano che, finalmente, si è dovuta fare contro una database di EST humani. Questa validazione per BLAST a un senso perchè si è confermato, a traverso una comparazione di sequenze animali che contenevano IRE, che la conservazione di identità di sequenza è molto più grande (piů del 90%), fra specie per lo stesso mRNA che fra diversi mRNA dentro la stessa specie (36-85% identitŕ).
I risultati ottenuti dopo tutto questo processo non sono stati molto significativi, come abbiamo detto, anche se è stato per motivi esterni a noi. Tuttavia, dobbiamo pensare che questa è soltanto una piccola parte del totale del lavoro, i cui risultati finali si mostrano in questo link.
2-03-2003 E-mail: anuky4@hotmai.com ; yurcelay@hotmail.com
Con questo modello descrito in la bibliografia [1], come mostra il disegno, abbiamo sperato di trovare cinque acoppiamenti esatti nella "stem" superiore e da due a otto acoppiamenti nella "stem" inferiore. In piů, abbiamo accettato gli acoppiamenti A-T e G-C, ed anche i G-T, perchè si è observato che, anche se questi ultimi non sono acoppiamenti classici, si trovano in un numero importante di strutture secondarie.
Questo modello, che noi abbiamo definito dai dati trovati negli articoli, è diverso dall'anteriore nel fatto che si permettono quattro o cinque acoppiamenti nella stemm superiore.
2. Esecuzione del programma "Scan_for_matches"
3. Traduzione dei risultati a linguaggio FASTA
4. BLAST contro genoma umano
5. Risultati
