Previo al inicio del análisis, se buscó información bibliográfica sobre las selenoproteínas y los diferentes métodos de estudio utilizados para su descubrimiento y descripción. Una vez asignada la especie problema, Trypanosoma brucei, se investigó acerca de la biología del organismo y la patología que produce.
El paso siguiente consistió en adquirir los archivos necesarios para iniciar la búsqueda de las selenoproteínas y sus homólogas en T. brucei.
El genoma del organismo y los archivos que forman la base de datos necesaria para poder ejecutar el programa TBLASTN desde la ventana terminal, fueron adquiridos mediante un link proporcionado por el profesorado de la asignatura de Bioinformática: (~/.novell-servers/REC-C1AU.UPF.ES/PUBLIC/FOLDERS/3361/12306-1/disc8/genomes/T.brucei).
Las secuencias de las selenoproteínas en formato fasta se obtuvieron en la base de datos Seleno DB. Nos centramos en las selenoproteínas descritas en Homo sapiens, ya que es la especie que mayor número de selenoproteínas posee. Se analizaron un total de 37 proteínas incluyendo las selenoproteínas descritas y sus homólogas con cisteína u otro aminoácido. Cada familia se analizó por separado llevando a cabo el mismo protocolo.
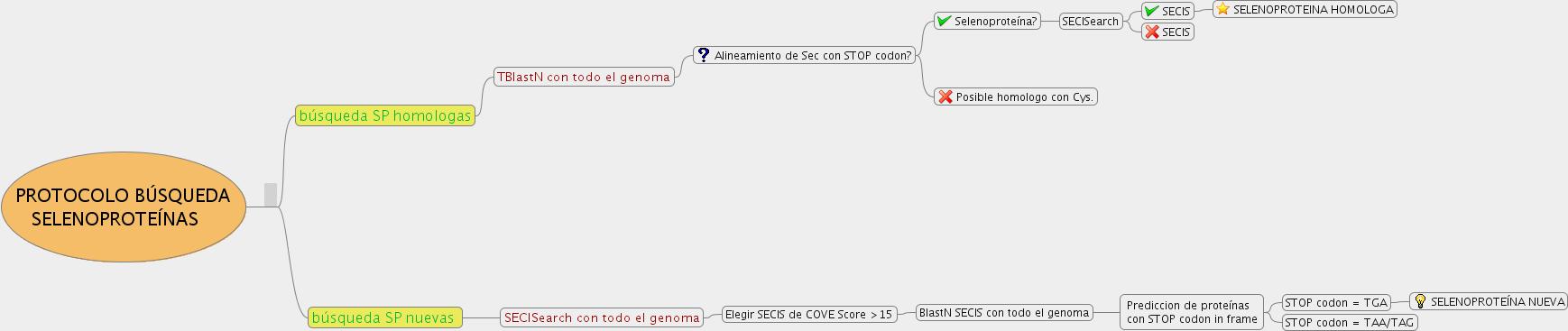
La búsqueda inicial de selenoproteínas y proteínas homólogas se realizó mediante el programa TBLASTN. El programa se ejecutó con los parámetros establecidos por defecto. Para hacer una valoración de los alineamientos obtenidos, seleccionamos aquellos que presentaron un e-value menor que 0,1.En los casos en que este parámetro no fue suficiente, también se consideraron los valores del bit score.
Los programas TblToFasta.pl y FastaToTbl.pl se utilizaron para extraer las subsecuencias de los cromosomas que abarcaban los hits de interés. Posteriormente, estas subsecuencias obtenidas se alinearon con la proteína que había generado cada hit mediante el programa GeneWise. Este progama nos permitió hacer la predicción de la secuencia proteíca y genómica.
La búsqueda de todos los posibles SECIS en el genoma de T.brucei se realizó mediante el programa SECISearch. El programa se ejecutó con todos los parámetros por defecto:
El COVE score mínimo recomendado por el propio programa es de 15, por lo tanto en un primer momento sólo elegimos los SECIS que poseían un puntaje mayor a éste. Debido a que el SECIS de la SelT de T.brucei presenta un COVE score de 8,66 se amplió el rango de búsqueda eligiendo también aquellos que presentaban un COVE score mayor a 8.
La secuencia de los SECIS seleccionados se pasó a DNA (cambio de los uracilos por timinas) mediante el comando reemplazar del editor de texto del paquete OpenOffice. Las secuencias obtenidas se pusieron en formato fasta y se realizó un BLASTN contra todo el genoma.
De los resultados del BLASTN sólo se escogieron aquellos que presentaban 100% de identidad con la secuencia en el alineamiento y que tenían un e-value menor que 0, dejando sólo uno o dos hits por cada secuencia buscada.
Los alineamientos del BLASTN se utilizaron para calcular las coordenadas de las subsecuencias para posteriormente buscar las potenciales selenoproteínas. Las coordenadas se obtuvieron restando 3000 nucleótidos al s.start que indicaba el inicio del alineamiento. También se calculó el largo de la secuencia de los SECIS buscados para añadirlas al largo total de cada subsecuencia.
La extracción de los cromosomas y de las subsecuencias se realizó igual que en apartados anteriores mediante los programas FastaToTbl.pl y TblToFasta.pl.
El siguiente paso fue buscar las proteínas que comenzaban con metionina y tenían un codón STOP in frame. Para ello se obtuvieron las seis posibles pautas de lectura de cada subsecuencia con el comando fastatranslate y se resaltaron aquellas posibles proteínas que cumplían las condiciones anteriores. El comando fastarevcomp se utilizó para encontrar la secuencia reversa complementaria de algunas subsecuencias.
Por último se utilizó el programa GeneWise para realizar un alineamiento de las posibles proteínas encontradas en las pautas de lectura y la subsecuencia inicial de cada una para comprobar si el codón STOP codon in frame correspondía a un TGA (codificante también para la selenocisteína).
| <- Introducción | arriba | Resultados -> |