
COMPUTATIONAL GENOMIC STUDY OF AML1-MDS1-EVI1
Cristina del Barrio Gascón (cristina.delbarrio01@upf.edu) and Belén García Izquierdo (belen.garcia02@upf.edu)
Facultat de Ciències de la Salut i de la Vida
Universitat Pompeu Fabra
Protein number 7 is a three-protein fusion. It is composed by evi1, mds1 and aml1. EVI1 and MDS1 genes are located at chromosome 3, whereas the AML1 gene maps at chromosome 21.
The t(3;21)(q26;q22) translocation, which is one of the chromosomal abnormalities found in blastic crisis of chronic myelocytic leukemia (CML), is thought to play an important role in the leukemic progression of CML to an acute blastic crisis phase.
In cells with t(3;21) translocation, the observed fusion transcripts are AML1-MDS1 and AML1-MDS1-EVI1, being this last our fusion protein. This translocation results in the formation of a new class of chimeric transcription factor which could contribute to the leukemic progression of CML through interference with cell growth and differentiation.
In this Bioinformatics project we have analyzed different parameters for each of the 3 proteins which configure our fusion protein. These parameters are: genomic structure, homology among other species, gene expression in different tissues, promoter regions and gene function. With all this information, and also thanks to the consulted literature, we have been able to describe AML1-MDS1-EVI1 (AME) structure and mechanism.
Defined karyotypic abnormalities are associated with specific subtypes of human leukemias. Molecular characterization of these abnormalities, especially reciprocal translocations, has shown that the chromosomal abnormalities are implicated in leukemogenesis by altering the function of genes located at or near the translocation breakpoint. Important mechanisms in leukemogenesis are the inappropriate expression of transcription factors or the creation of fusion mRNAs and fusion transcription factor proteins by chromosomal translocation.
Chronic myelocytic leukemia (CML) is a clonal disorder of pluripotent haematopoietic stem cells usually with a biphasic clinical course. An initial chronic phase characterized by leukocytosis with maturation is usually followed by acceleration of the disease and eventually by an acute blastic crisis phase characterized by cellular proliferation, maturation arrest and karyotypic clonal evolution.
A reciprocal translocation, t(9;22)(q34;q11), is the hallmark of the chronic phase of CML. It is known that additional chromosomal abnormalities to the t(9;22) translocation appear in or before the blastic crisis phase of CML. The t(3;21)(q26;q22) translocation is one of these additional chromosomal abnormalities. Because this structural change occurs prior or near the moment of blastic crisis development, it could play a causative role in blastic crisis of CML and also in myelodysplastic syndrome (MDS) -derived leukemia.
In cells with t(3;21) translocation, the resulting fusion transcripts are AML1-MDS1 or AML1-MDS1-EVI1. As we can see in our protein 7, the t(3;21) translocation leads to AML1-MDS1-EVI1 fusion protein.
AML1 is located at 21q22.3. It is essential for haematopoietic cell development in fetal liver and its lineage-specific differentiation in adult. AML1 is expressed in all tissues examined except brain and heart. The highest levels are found in thymus, bone marrow and peripheral blood.
EVI1 maps 3q26.2. Unlike AML1, EVI1 is barely expressed in normal haematopoietic cells, but it is over expressed in chronic myelocytic leukemia in blastic crisis and myelodysplastic syndrome-derived leukemia.
MDS1 is located at 3q26.2. MDS1 is a separate gene which is located upstream of EVI1. It codifies for a transcription factor of unknown function. The fusion protein MDS1/EVI1 is also expressed normally through intergenic splicing. MDS1/EVI1 contains the PR domain that is about 40% homologous to the N-terminus of retinoblastoma-binding protein, RIZ (retinoblastoma interacting zinc-finger).
Because it is very rare that de novo acute leukemia carries the t(3;21) translocation, the appearance of this chromosomal abnormality may be a key event when haematological disorders arise from haematopoietic stem cells.
The present project consisted on the analysis of different aspects of a given protein. Our group was assigned protein number 7.
At first, we thought that protein 7 was composed by only 2 proteins: AML1 and EVI1. There was an unidentified short sequence between both of them which was afterwards determined as a third protein: MDS1. As our given protein was an AML1-MDS1-EVI1 fusion, we analyzed each of the 3 proteins separately.
The points to cover in the whole study were the following:
1. Description of the genomic structure of the 3 genes which codified AML1, MDS1 and EVI1 proteins, respectively.
- Chromosomic localization, number of coding exons, number of non-coding exons and number of transcripts associated to each of the 3 genes.
- Determination of the fact if the transcripts associated to each gene included groups of different exons due to alternative splicing. If so, enumeration of the exons which were correlated to the different isoforms, indicating whereas the isoform difference did affect a coding or a non-coding portion of the gene. In the coding case, resolution if the reading frame was conserved or, on the contrary, if it produced a different protein.
Firstly, we entered NCBI and performed tblastn of our original sequence. We chose the value with the highest score and it was our fusion protein. We entered the link and confirmed it was from Homo sapiens. The first 178 nucleotides were part of Aml1 and the rest until 1395 formed part of Evi1.
We read an article which was there cited. It explained the same as in NCBI, but it claimed that the part that we now know to be Mds1 was a fragment of an Evi1 coding region. We did not understand why it was transcribed if it was supposed to be non-coding.
Afterwards, we entered Ensembl and pasted our fusion protein. It appeared to be just Evi1. Then we did it separately, but we could not get to know what the part between Aml1 and Evi1 was. When analyzing this in-between fragment using Ensembl, only one part was shown to be a fragment of Mds1, but we still did not know what the rest was.
Finally, we found out that the part which was between Mds1 and Evi1 was an Evi1 non-coding fragment.
2. Study of each gene homology in other species.
- Elaboration of a table of orthologue species per each gene.
- Determination of the identity percentage in the corresponding alignments.
We got the orthologues for AML1 and MDS1 from Ensembl. We could not get the orthologues for EVI1
from Ensembl, so they were obtained from UCSC Genome Browser linking to NCBI.
3.Description of the gene expression in different tissues or cell types.
It was done it in 2 ways:
- We went to UCSC Genome Browser and, after doing BLAST, we observed each gene the expression in Gene Sorter. We also looked at EST, but this tool was not useful as we were asked for a tissue.
- We performed VisiGene, from UCSC Genome Browser. Anyway, there only appeared images from Mus musculus, which was not practical as we had a Homo sapiens sequence.
4. Description of the promoter region of each of the 3 genes.
- Extraction of the promoter region of each gene, formed by 1 kb upstream the transcription start site (TTS) and 100 bp downstream the TSS.
- Selection of a cluster of transcription factors (TFs) that putatively binded our promoter region and thus could be regulating the gene expression in each of our 3 genes.
This selection was performed in two ways:
Designing a Perl program which was developed following the given instructions.
Using the web server of PROMO program.
Inside Ensembl, for each of our transcripts, we went to exon information and we flanked our sequence at either end of transcript.
In PROMO we pasted our 3 sequences separately and we filtered our TF results (5% maximum matrix dissimilarity rate) per each gene. We saved the data file in a .txt format. We transferred it to an Excel file and we performed a selection of TFs based on those parameters:
- Dissimilarity, Re query and Re equally: The lower the better.
- String: The longer the better.
The selected TFs should coincide with the given TFs in the Perl program instructions. Therefore, we displayed a further selection of our TFs. We chose the TFs which had a Re equally no higher than 0.09. As Re equally parameter is similar to p-value parameter, we compared our last TFs selection with the TFs from the Perl matrices.
5. Study of the gene function in each gene.
- Using Gene Ontology database, with the GO term annotation associated to each of the 3 genes.
- From the overall previous analysis and the research in the available literature (PubMed, basically).
After performing our computational analysis using different databases and software, we show the obtained results, which are divided into several points:
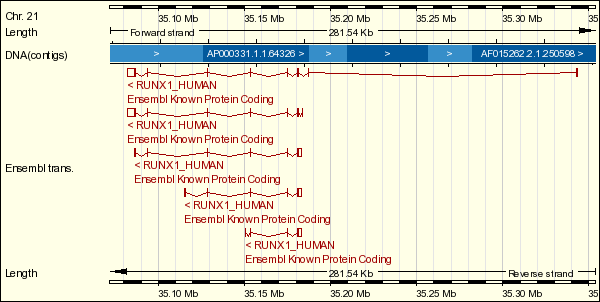
If we search for AML1 in the Ensembl database 5 transcripts appear, whereas in Genome Browser only 2 transcripts do appear. Therefore, we decided to base our analysis in Ensembl.
MASDSIFESFPSYPQCFMRECILGMNPSRDVHDASTSRRFTPPSTALSPGKMSEALPLGAPDAGAALAGKLRSGDRSMVEVLADHPGELVRTDSPNFLCSVLPTHWRCNKTLPIAFKVVA LGDVPDGTLVTVMAGNDENYSAELRNATAAMKNQVARFNDLRFVGRSGRGKSFTLTITVFTNPPQVATYHRAIKITVDGPREPRRHRQKLDDQTKPGSLSFSERLSELEQLRRTAMRVSP HHPAPTPNPRASLNHSTAFNPQPQSQMQDTRQIQPSPPWSYDQSYQYLGSIASPSVHPATPISPGRASGMTTLSAELSSRLSTAPDLTAFSDPRQFPALPSISDPRMHYPGAFTYSPTPV TSGIGIGMSAMGSATRYHTYLPPPYPGSSQAQGGPFQASSPSYHLYYGASAGSYQFSMVGGERSPPRILPPCTNASTGSALLNPSLPNQSDVVEAEGSHSNSPTNMAPSARLEEAVWRPY
In order to make out whether the guideline of reading was or not maintained, we counted the number of coding nucleotides from the gene sequence. If it was a 3 multiple, it was considered that the pattern of [3 nucleotides : 1 codon] was conserved. Therefore, the guideline reading was maintained. At a protein level, we compared a large sequence of amino acids between transcripts. If the same fragment was present, it was clear that the guideline reading was conserved.
In this case, the same guideline of reading is conserved.
MPAAPRGPAQGEAAARTRSRDASTSRRFTPPSTALSPGKMSEALPLGAPDAGAALAGKLRSGDRSMVEVLADHPGELVRTDSPNFLCSVLPTHWRCNKTLPIAFKVVALGDVPDGTLVTV
MAGNDENYSAELRNATAAMKNQVARFNDLRFVGRSGRGKSFTLTITVFTNPPQVATYHRAIKITVDGPREPRRHRQKLDDQTKPGSLSFSERLSELEQLRRTAMRVSPHHPAPTPNPRAS
LNHSTAFNPQPQSQMQDTRQIQPSPPWSYDQSYQYLGSIASPSVHPATPISPGRASGMTTLSAELSSRLSTAPDLTAFSDPRQFPALPSISDPRMHYPGAFTYSPTPVTSGIGIGMSAMG
SATRYHTYLPPPYPGSSQAQGGPFQASSPSYHLYYGASAGSYQFSMVGGERSPPRILPPCTNASTGSALLNPSLPNQSDVVEAEGSHSNSPTNMAPSARLEEAVWRPY
- Comparison between ENST00000300305 transcript (8 exons) and ENST00000325074 transcript (7 exons):
ENST00000325074 transcript has one exon less than ENST00000300305 transcript. When comparing the nucleotidic sequence we can see that the last 6 exons are the same for both transcripts. However, when looking at the 1st exon from ENST00000325074 transcript, we realize it does not match with the 2nd exon, actually it belongs to an intronic sequence from ENST00000300305 transcript.
The same guideline of reading is conserved.
MRIPVDASTSRRFTPPSTALSPGKMSEALPLGAPDAGAALAGKLRSGDRSMVEVLADHPGELVRTDSPNFLCSVLPTHWRCNKTLPIAFKVVAL GDVPDGTLVTVMAGNDENYSAELRNATAAMKNQVARFNDLRFVGRSGRGKSFTLTITVFTNPPQVATYHRAIKITVDGPREPRNSLTWPRYPHI
- Comparison between ENST00000342083 transcript (4 exons) and ENST00000300305 transcript (8 exons):
ENST00000342083 transcript is shorter than ENST00000300305 transcript, as it only includes 4 exons. When looking at the nucleotidic sequence we observe that both 4th and 5th exons from ENST00000300305 transcript match exactly with the 2nd and the 3rd exons from ENST00000342083 transcript. With regard to the 1st exon from ENST00000342083 transcript, it belongs to the 3rd exon from ENST00000300305 transcript. Regarding the last exon (the 4th one) we can see that it does not match with the 6th exon because the sequence belongs to an intronic region which, after the alternative splicing, becomes a coding region.
The same guideline of reading is conserved.
MRIPVDASTSRRFTPPSTALSPGKMSEALPLGAPDAGAALAGKLRSGDRSMVEVLADHPG ELVRTDSPNFLCSVLPTHWRCNKTLPIAFKVVALGDVPDGTLVTVMAGNDENYSAELRNA TAAMKNQVARFNDLRFVGRSGRGKSFTLTITVFTNPPQVATYHRAIKITVDGPREPRRHR QKLDDQTKPGSLSFSERLSELEQLRRTAMRVSPHHPAPTPNPRASLNHSTAFNPQPQSQM QDTRQIQPSPPWSYDQSYQYLGSIASPSVHPATPISPGRASGMTTLSAELSSRLSTAPDL TAFSDPRQFPALPSISDPRMHYPGAFTYSPTPVTSGIGIGMSAMGSATRYHTYLPPPYPG SSQAQGGPFQASSPSYHLYYGASAGSYQFSMVGGERSPPRILPPCTNASTGSALLNPSLP NQSDVVEAEGSHSNSPTNMAPSAWRRPSRRDPGPWARPPSWGRGRPTARISL
- Comparison between ENST00000344691 transcript (8 exons) and ENST00000300305 transcript (8 exons):
We can see that ENST00000344691 transcript contains 8 exons just like ENST00000300305 transcript. If we look the nucleotidic sequence in detail, we observe that the 2nd, 3rd, 4th and 5th exons from ENST00000344691 transcript with the 4th, 5th, 6th and 7th exons from ENST00000300305 transcript. Regarding the 6th exon from ENST00000344691 transcript, it is the same as the 8th exon from ENST00000300305 transcript but, in this case, it ends in a few prior positions. With regard to the 1st exon we observe again that it belongs to the 3rd exon from ENST00000300305 transcript.
The same guideline of reading is conserved.
MRIPVDASTSRRFTPPSTALSPGKMSEALPLGAPDAGAALAGKLRSGDRSMVEVLADHPGELVRT
DSPNFLCSVLPTHWRCNKTLPIAFKVVALGDVPDGTLVTVMAGNDENYSAELRNATAAMKNQVAR
FNDLRFVGRSGRGKSFTLTITVFTNPPQVATYHRAIKITVDGPREPRRHRQKLDDQTKPGSLSFS
ERLSELEQLRRTAMRVSPHHPAPTPNPRASLNHSTAFNPQPQSQMQEEDTAPWRC
- Comparison between ENST00000358356 transcript (5 exons) and ENST00000300305 transcript (8 exons), being this last our transcript:
We realize that this transcript is formed only by 5 exons. When looking at the nucleotidic sequence we can see that the 2nd, 3rd and 4th exons from ENST00000358356 transcript match with the 4th, 5th and 6th exons from ENST00000300305 transcript. The 5th exon from ENST00000358356 transcript is different from the 7th exon from ENST00000300305 transcript as these nucleotides are found at intronic region. The 1st exon from ENST00000358356 transcript, as it has been mentioned before, belongs to the 3rd exon from ENST00000300305 transcript.
The same guideline of reading is conserved.
If we perform an alignment of the proteins that are derived from the 5 transcripts, which were obtained using Clustalw program, we can see a high aminoacidic conservation between the different transcripts. What is different among them is the ending place where the sequence finishes: it can be longer or shorter depending on the transcript. In order to see the obtained alignment click here
With the information showed above we can conclude that, although alternative splicing produces different transcripts, the resulting protein conserves a whole part of the sequence. Thus, the guideline of reading is maintained in all transcripts. Therefore, the only difference is the length of the part which is before and after the conserved fragment.
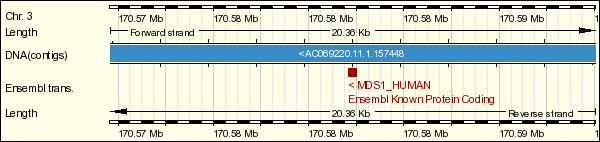
The gathered data displays information of the different MDS1 transcripts obtained from Ensembl.
Due to the absence of alternative splicing, MDS1 gene only has this transcript, meaning there are no Mds1 isoforms.
SSYLFTDNECVYGNYPEIPLEEMPDADGVASTPSLNIQEPCSPATSSEAFTPKEGSPYKA
PIYIPDDIPIPAEFELRESNMPGAGLGIWTKRKIEVGEKFGPYVGEQRSNLKDPSYGWEVR
As this transcript was not very similar to our given sequence, we chose the OTTHUMG00000137072 Vega transcript, which was much more similar to our sequence.
The Vega transcript is a product of the BCM:MDS1 gene. Like above, the Vega gene only has this transcript, meaning again there is not any Mds1 isoforms.
Transcript OTTHUMT00000267016:
MRSKGRARKLATNNECVYGNYPEIPLEEMPDADGVASTPSLNIQEPCSPATSSEAFTPKEGSPYKAPIYIPDDIPIPAEFELRESN
MPGAGLGIWTKRKIEVGEKFGPYVGEQRSNLKDPSYGWEVHLPRSRRVSVHSWLYLGKRSSDVGIAFSQADVYMPGLQCAFLS
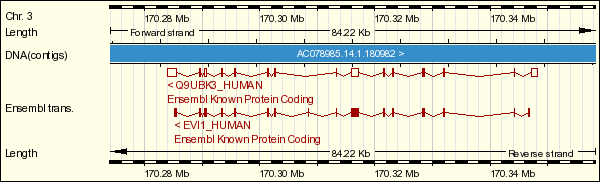
Because of alternative splicing, EVI1 gene has 2 transcripts which correspond to 2 different Evi1 isoforms.
MKSEDYPHETMAPDIHEERQYRCEDCDQLFESKAELADHQKFPCSTPHSAFSMVEEDFQQKLESEN
DLQEIHTIQECKECDQVFPDLQSLEKHMLSHTEEREYKCDQCPKAFNWKSNLIRHQMSHDSGKHYE
CENCAKVFTDPSNLQRHIRSQHVGARAHACPECGKTFATSSGLKQHKHIHSSVKPFICEVCHKSYT
QFSNLCRHKRMHADCRTQIKCKDCGQMFSTTSSLNKHRRFCEGKNHFAAGGFFGQGISLPGTPAMD
KTSMVNMSHANPGLADYFGANRHPAGLTFPTAPGFSFSFPGLFPSGLYHRPPLIPASSPVKGLSST
EQTNKSQSPLMTHPQILPATQDILKALSKHPSVGDNKPVELQPERSSEERPFEKISDQSESSDLDD
VSTPSGSDLETTSGSDLESDIESDKEKFKENGKMFKDKVSPLQNLASINNKKEYSNHSIFSPSLEE
QTAVSGAVNDSIKAIASIAEKYFGSTGLVGLQDKKVGALPYPSMFPLPFFPAFSQSMYPFPDRDLR
SLPLKMEPQSPGEVKKLQKGSSESPFDLTTKRKDEKPLTPVPSKPPVTPATSQDQPLDLSMGSRSR
ASGTKLTEPRKNHVFGGKKGSNVESRPASDGSLQHARPTPFFMDPIYRVEKRKLTDPLEALKEKYL
RPSPGFLFHPQFQLPDQRTWMSAIENMAEKLESFSALKPEASELLQSVPSMFNFRAPPNALPENLL
RKGKERYTCRYCGKIFPRSANLTRHLRTHTGEQPYRCKYCDRSFSISSNLQRHVRNIHNKEKPFKC
HLCDRCFGQQTNLDRHLKKHENGNMSGTATSSPHSELESTGAILDDKEDAYFTEIRNFIGNSNHGS
QSPRNVEERMNGSHFKDEKALVTSQNSDLLDDEEVEDEVLLDEEDEDNDITGKTGKEPVTSNLHEG
NPEDDYEETSALEMSCKTSPVRYKEEEYKSGLSALDHIRHFTDSLKMRKMEDNQYSEAELSSFSTS
HVPEELKQPLHRKSKSQAYAMMLSLSDKESLHSTSHSSSNVWHSMARAAAESSAIQSISHV
ILDEFYNVKFCIDASQPDVGSWLKYIRFAGCYDQHNLVACQINDQIFYRVVADIAPGEEL
LLFMKSEDYPHETMAPDIHEERQYRCEDCDQLFESKAELADHQKFPCSTPHSAFSMVEED
FQQKLESENDLQEIHTIQECKECDQVFPDLQSLEKHMLSHTEEREY
Comparison between ENST00000343372 transcript (15 exons) and ENST00000264674 transcript (16 exons):
We can see that ENST00000343372 transcript has one exon less than ENST00000264674 transcript. When looking at the nucleotidic sequence, we observe that the exons are formed by the same nucleotides. However, in ENST00000343372 transcript, the majority of nucleotides become non-coding, leading to a shorter protein. The missing exon is the 9th one.
The same guideline of reading is conserved.
Until here, we have studied our 3 proteins separately. Now, we are going to analyze our fusion protein (AME). The given sequence is:
MRIPVDASTSRRFTPPSTALSPGKMSEALPLGAPDAGAALAGKLRSGDRSMVEVLADHPGELVRTDSPNF
LCSVLPTHWRCNKTLPIAFKVVALGDVPDGTLVTVMAGNDENYSAELRNATAAMKNQVARFNDLRFVGRS
GRGKSFTLTITVFTNPPQVATYHRAIKITVDGPREPR
NNECVYGNYPEIPLEEMPDADGVASTPSLNIQE
PCSPATSSEAFTPKEGSPYKAPIYIPDDIPIPAEFELRESNMPGAGLGIWTKRKIEVGEKFGPYVGEQRS
NLKDPSYGWEILDEFYNVKFCIDASQPDVGSWLKYIRFAGCYDQHNLVACQINDQIFYRVVADIAPGEEL
LLFMKSEDYPHETMAPDIHEERQYRCEDCDQLFESKAELADHQKFPCSTPHSAFSMVEEDFQQKLESEND
LQEIHTIQECKECDQVFPDLQSLEKHMLSHTEEREYKCDQCPKAFNWKSNLIRHQMSHDSGKHYECENCA
KVFTDPSNLQRHIRSQHVGARAHACPECGKTFATSSGLKQHKHIHSSVKPFICEVCHKSYTQFSNLCRHK
RMHADCRTQIKCKDCGQMFSTTSSLNKHRRFCEGKNHFAAGGFFGQGISLPGTPAMDKTSMVNMSHANPG
LADYFGANRHPAGLTFPTAPGFSFSFPGLFPSGLYHRPPLIPASSPVKGLSSTEQTNKSQSPLMTHPQIL
PATQDILKALSKHPSVGDNKPVELQPERSSEERPFEKISDQSESSDLDDVSTPSGSDLETTSGSDLESDI
ESDKEKFKENGKMFKDKVSPLQNLASINNKKEYSNHSIFSPSLEEQTAVSGAVNDSIKAIASIAEKYFGS
TGLVGLQDKKVGALPYPSMFPLPFFPAFSQSMYPFPDRDLRSLPLKMEPQSPGEVKKLQKGSSESPFDLT
TKRKDEKPLTPVPSKPPVTPATSQDQPLDLSMGSRSRASGTKLTEPRKNHVFGGKKGSNVESRPASDGSL
QHARPTPFFMDPIYRVEKRKLTDPLEALKEKYLRPSPGFLFHPQMSAIENMAEKLESFSALKPEASELLQ
SVPSMFNFRAPPNALPENLLRKGKERYTCRYCGKIFPRSANLTRHLRTHTGEQPYRCKYCDRSFSISSNL
QRHVRNIHNKEKPFKCHLCDRCFGQQTNLDRHLKKHENGNMSGTATSSPHSELESTGAILDDKEDAYFTE
IRNFIGNSNHGSQSPRNVEERMNGSHFKDEKALVTSQNSDLLDDEEVEDEVLLDEEDEDNDITGKTGKEP
VTSNLHEGNPEDDYEETSALEMSCKTSPVRYKEEEYKSGLSALDHIRHFTDSLKMRKMEDNQYSEAELSS
FSTSHVPEELKQPLHRKSKSQAYAMMLSLSDKESLHSTSHSSSNVWHSMARAAAESSAIQSISHV
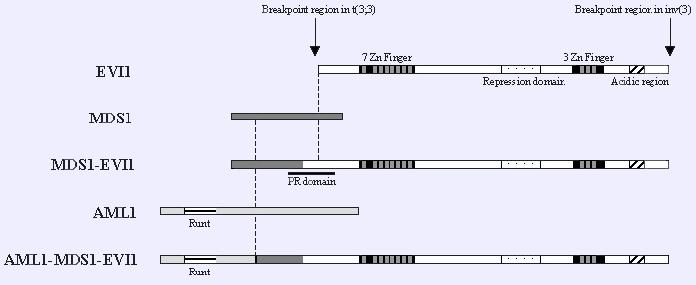
The purple part is Aml1, the green part is Mds1, the pink part is the Evi1 non-coding part and the blue part is the Evi1 coding part.
If we pay attention to the structure of our fusion protein, we can see that the Aml1 part which is present in the fusion protein includes the first 3 exons from the ENST00000342083 transcript.
Referring to MDS1, the transcript which is considered to form part of the fusion protein is OTTHUMG00000137072 Vega transcript. In the fusion protein, only the 3rd exon from MDS1 is included. It is important to mention that the N aminoacid (shown in red in the sequence) is encoded by the last A nucleotide of the 3rd exon from AML1 and the 2 first nucleotides (A and T) of the 3rd exon from MDS1.
The pink sequence shows the 2nd exon and a part of the 3rd exon, which form part of our EVI1 ENST00000264674 transcript. This fragment is non-coding in EVI1, but it turns to be a coding sequence in our fusion protein.
The blue sequence corresponds to the coding part of the 3rd exon from EVI1, and the rest of the EVI1 coding sequence. It means that the blue part starts at the EVI1 transcription start site and includes the rest of the gene coding sequence, which is going to produce the whole evi1 protein.
AML1, MDS1 and EVI1 are transcribed in reverse direction (from telomere to centromere). After the t(3;21)(q26;q22) translocation the 3 genes get transcribed one after the other following this order: AML1-MDS1-EVI1. Then, MDS1 and EVI1 become regulated by the AML1 gene promoter.
In this part of our project we perform a study of the gene homology -for each of our 3 genes- in different species. Orthologues for AML1 and MDS1 were obtained from Ensembl, whereas we got the orthologues for EVI1 from UCSC Genome Browser and NCBI.
There were organism which had more than one entry displayed. In those cases, we selected the one showing the higher % target identity. Anyway, we show all the results without rejecting any low %identity value. In the type column we distinguish the many and one types. The first type means that sequences from many organisms had been compared, whereas the one type means that only the sequence from a single organism has been aligned. The last column presents the gene identifiers to enter the respective database if further information is required.
Besides, we have performed an alignment of all the aminoacidic sequences from the different human orthologues which are shown in the tables. The aminoacid sequences were obtained from Ensembl and Genome Browser. We made an alignment and a tree per each of the 3 proteins using ClustalW. We used ClustalW in order to show the homology between species in a graphical way and also to corroborate the table results.
The %identity results for each gene are resumed in the following tables:
Aedes aegypti |
|||
Anopheles gambiae |
|||
Bos taurus |
|||
Caenorhabditis elegans |
|||
Canis familiaris |
|||
Cavia porcellus |
|||
Ciona intestinalis |
|||
Ciona savignyi |
|||
Dasypus novemcinctus |
|||
Drosophila melanogaster |
|||
Echinops telfairi |
|||
Felis catus |
|||
Gallus gallus |
|||
Gasterosteus aculeatus |
|||
Loxodonta africana |
|||
Macaca mulatta |
|||
Monodelphis domestica |
|||
Mus musculus |
|||
Ornithorhynchus anatinus |
|||
Oryctolagus cuniculus |
|||
Oryzias latipes |
|||
Pan troglodytes |
|||
Rattus norvegicus |
|||
Takifugu rubripes |
|||
Tetraodon nigroviridis |
|||
Tupaia belangeri |
|||
Xenopus tropicalis |
|||
Analyzing the data, we find out that the two species with the highest homology with Homo sapiens are Pan troglodytes(100%) and Macaca mulatta(99%). It is a reasonable observation due to the fact that those are the closer species in the filogenetic scale. On the other hand, we point at the case of C. elegans, which shows a very low % target identity. This correlates with what was described above because, as C. elegans is more separated from humans in the speciation time, we observe a very low % target identity. Consequently, we realize that the closer the species are in the evolutive tree, higher is the %identity they share.

When looking at the tree, Pan troglodytes appers, as it was expected, the closest specie to human. Unexpectedly, Macaca mulatta is placed further than it should be, compared to what the previous %homology displayed. The reason we suggest to explain this fact is that maybe the Canis familiaris is to blame. In this kind of alignment trees, dog sequences are usually removed as they are not that reliable (because they are humanized sequences). As we have conserved the specie in our tree, it may have disarrenged the %identity between macaque and human.
Bos taurus |
|||
Canis familiaris |
|||
Cavia porcellus |
|||
Dasypus novemcinctus |
|||
Erinaceus europaeus |
|||
Felis catus |
|||
Gasterosteus aculeatus |
|||
Loxodonta africana |
|||
Macaca mulatta |
|||
Monodelphis domestica |
|||
Mus musculus |
|||
Ornithorhynchus anatinus |
|||
Rattus norvegicus |
|||
Tetraodon nigroviridis |
|||
Tupaia belangeri |
|||
Xenopus tropicalis |
|||
Analyzing the table, we can see that the species which share the highest homology with Homo sapiens are Macaca mulatta(99%) and Felis catus(95%). It is a reasonable observation due to the fact that the macaque and the cat are the closer species to the human in the evolutive tree.
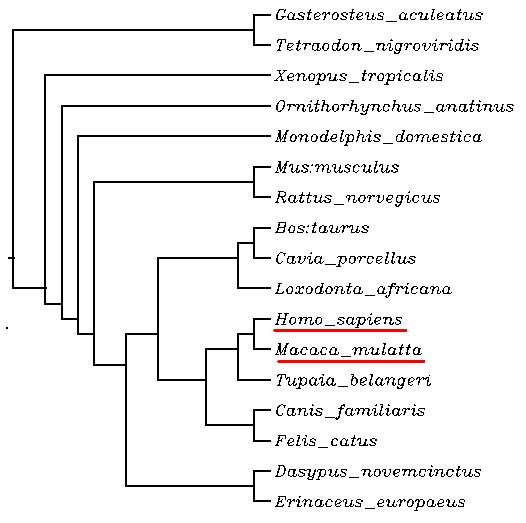
If we look at the 2 underlined species in the ClustalW tree, we can corroborate that Macaca mulatta is the closest species to humans.
Pan troglodytes |
||
Canis familiaris |
||
Mus musculus |
||
Rattus norvegicus |
||
Gallus gallus |
||
Drosophila melanogaster |
||
Anopheles gambiae |
||
Analyzing the data, we can see that the species which share the highest homology with Homo sapiens are Pan troglodytes(99.4%) and Canis familiaris(96.5%). It is a reasonable observation due to the fact that both chimpanzee and dog are the closest species to human in terms of phylogeny.
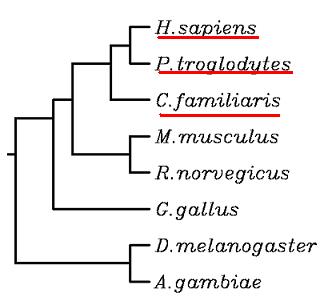
When looking at the 3 underlined species in the tree, we can see that Pan troglodytes and Canis familiaris are the closest species to Homo sapiens. So, this corroborates the table results which we have explained above.
The following figure was obtained from Gene Sorter. It shows AML1 expression in different tissues and cell types:
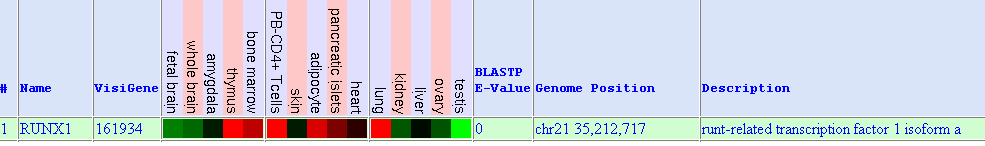
Red colour means high gene expression whereas green colour shows low expression. We observe high expression in: thymus, bone marrow, PB-CD4+ T cells, adipocyte, pancreatics islets and lung. Low expression is observed in fetal brain, whole brain, kidney, ovary and testis. Appart from this graphic, when performing further insight, we find out that Aml1 is widely expressed, including haematopoietic cells at various stages of differenciation. This fact leads us to its significant role in haematopoiesis.
The following figure was obtained from Gene Sorter. It shows MDS1 expression in different tissues and cell types:
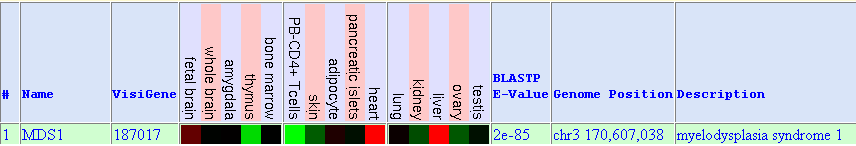
Red colour means high gene expression whereas green colour shows low expression. We observe high expression in: fetal brain, heart and liver. Low expression is observed in thymus, PB-CD4+ T cells, skin, kidney and ovary. In further approaches, we have realized that Mds1 follows the same pattern of expression in adult tissues than Evi1 (which is described below).

Red colour means high gene expression whereas green colour shows low expression. We observe high expression in: skin, pancreatic islets, lung and kidney. Low expression is observed in whole brain, amygdale, thymus, PB-CD4+ T cells, adipocyte, heart, liver, ovary and testis. In some articles, it refers to the fact that Evi1 is less expressed in skeletal muscle.
In order to widen the knowledge related to gene expression and also to contrast some of the information which we found to be contradictory, we consulted the literature and here we show more information about this part:
AML1
AML1 is expressed mainly in the haematopoietic system as a complex pattern of mRNAs ranging in size between 2 and 8 kb. Several of these transcripts differ in their 5 and 3 UTRs. There are two distinct 5 UTR that exhibit different expression patterns in haematopoietic tissues and in response to mitogenic stimulation. These data raise the possibility that AML1 expression is transcriptionally regulated by two discrete promoter regions.
In several leukaemia-associated translocations AML1 is fused to other genes and transcription of the fused region is mediated by upstream sequences that normally regulate the expression of AML1. It is demonstrated that both upstream regions function as promoters in haematopoietic and non haematopoietic cell lines. The activity of both promoters is orientation-dependent and is enhanced, in a cell-type specific manner, by an heterologous enhancer sequence. These results indicate that additional control elements, either negative or positive, regulate the tissue-specific expression of AML1.
To show AML1 importance in the haematopoietic system, we include the figure below, which states the differences between wt Runx1 in blast cell colonies and KO Runx1 in the same cell type.
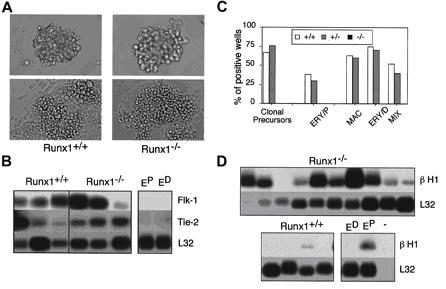
(A) Morphology of Runx1+/+ and Runx1/ blast cell colonies and the adherent and nonadherent cells generated from them following 2 days of culture (B) Gene expression analysis of adherent cells generated by individual blast colonies. Analysis was performed by the polyA+ global amplification method. Each line represents adherent cells derived from one blast colony. Control primitive erythroid (EP) and definitive erythroid (ED) samples are indicated. (C) Haematopoietic potential of the nonadherent population derived from expanded Runx1+/+ (+/+), Runx1+/ (+/), and Runx1/ (/) blast colonies. Nonadherent cells derived from individual colonies were harvested, and the cells were plated in conditions that support the growth of primitive and definitive colonies. The frequency of wells giving rise to total secondary colonies (clonal precursors), primitive erythroid colonies (ERY/P), or definitive haematopoietic colonies (MAC, macrophage; ERY/D, definitive erythroid; and MIX, multilineage) is represented. (D) H1 globin expression in nonadherent cells generated by individual colonies. Each line corresponds to the nonadherent cells from one expanded blast colony.
EVI1 and MDS1
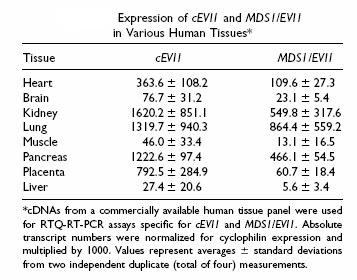
To compare the relative expression levels of cEVI1 and MDS1/EVI1 in normal human tissues, we show a RTQ-RT-PCR on a commercially available human cDNA panel. In brain, liver and muscle, both transcripts were of low abundance, but they were expressed at appreciable levels in heart, kidney, lung, pancreas and placenta. In most of the tissues, cEVI1 and MDS1/EVI1 were present at a ratio of approximately 3:1, with the exception of lung and placenta, where MDS1/EVI1 type transcripts accounted for as much as two thirds and as little as a tenth, respectively, of the cEVI1 mRNA.
Peripheral blood (PB) and/or bone marrow (BM) samples from six healthy controls and from 13 patients with myeloid leukemia and rearrangements of 3q26 were analyzed for expression of cEVI1 and MDS1/EVI1. Clinical characteristics, as well as expression values after normalization for cyclophilin, are summarized in the table below.
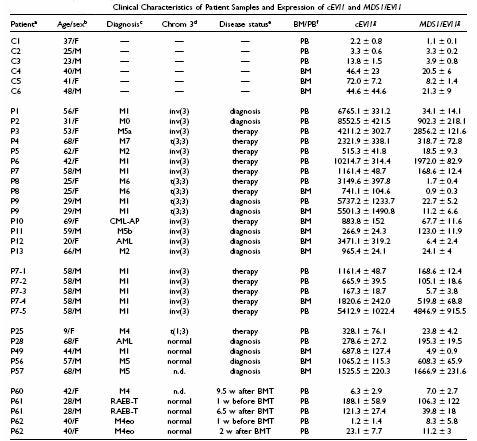
In the above table we can see:
C1 - C6, healthy controls; P1 - P13, patients with myeloid leukemia and rearrangements of chromosome band 3q26; P7(1) - P7(5), samples from patient P7 during the course of his disease; P25 - P57, patients with myeloid leukemia and cEVI1 overexpression in the absence of 3q26 aberrations; M, male; F, female.
M0 - M7, French American British (FAB) subtypes of acute myeloid leukemia (AML); M4eo, AML M4 with eosinophilia; AML, AML with unknown FAB type; CML-AP, accelerated phase of chronic myeloid leukemia; RAEB - T, refractory anemia with excess of blasts in transformation.
Cytogenetic status of chromosome 3. Normal, chromosome 3 not rearranged (other cytogenetic aberrations may be present); n.d., not determined. The unbalanced t(1;3) in patient P25 affected bands 3q23 - 25. Interphase FISH excluded involvement of the EVI1 locus.
For some of the patients from the P1 - P13 and the P25 - P57 groups, no sample from the time of diagnosis was available, but cytogenetics and/or morphological analysis had indicated the presence of aberrant cells in all of these. For P60 - P62, time before or after bone marrow transplantation (BMT) is indicated in weeks (w).
BM, bone marrow; PB, peripheral blood. Matched PB and BM samples of P8 and P9 were derived on the same days.
Expression values are absolute transcript numbers normalized for the amount of cyclophilin and multiplied by 1000. They represent averages-standard deviations from two independent duplicate (total of four) measurements.
For some patients, only samples obtained during therapy were available, but in all of these, cytogenetic and/or morphological analysis had indicated the persistence of the leukemic clone. In healthy donors, both cEVI1 and MDS1/EVI1 were expressed at very low levels, with transcription somewhat higher in BM than in PB.
cEVI1 levels were moderately to highly elevated in all patients with 3q26 rearrangements (Fig. 2A). In addition, it was also found a MDS1/EVI1 expression enhanced, compared to controls, in 8/9 PB and 2/6 BM samples from 3q26 patients (10/13 patients; Fig. 2B). In the majority of cases, MDS1/EVI1 was not induced as strongly as cEVI1, although it accounted for about half of the cEVI1 mRNA in patient samples P3 (PB) and P11 (BM).
In order to determine the TFs which binded our promoter sequences, we used the PROMO program. It is important to say that, due to the fact that there is more than one transcript for each gene, we have decided to base our analysis in the 3 transcripts which are considered the more similar to our fusion protein:
- AML1: transcript ENST00000342083. Promoter sequence
- MDS1: transcript OTTHUMG00000137072. Promoter sequence
- EVI1: transcript ENST00000264674. Promoter sequence
First of all, we have introduced the sequences in PROMO and have selected a 5% dissimilarity. Once the results have been obtained, we have done a selection of the TFs showing a Re equally inferior than 0.09. This parameter indicates the probability that a certain TF binds by chance a determinate nucleotide sequence with a 25% probability. So, we consider that Re equally values which are less than 0.09 show us that a certain TF will bind the promoter region. Because of that, its binding will not be produced by chance.
To follow with, we used a Perl program to determinate which of the TFs in a given file could bind our promoter regions in study. To that extent, we used a file which had a 13-TF matrix humanfactors_selectedmatrices and the 3 FASTA files from the promoter sequences cited before. The obtained results were the concrete positions where the different TFs binded each of the promoter sequences, and also the maximum score and its p-value. This p-value indicates the probability of wrongly refusing the initial hypothesis that the factor does not bind the sequence. Low values indicate that the identified binding is difficult to be observed by chance and, therefore, it is reasonable to think that the TF is actually binding the promoter region.
After using PROMO, we built a table with all the TFs ordered in an increasing Re equally order, until 0.09 (maximum). In the table we can see that, in the case of AML1, 12 TFs appear. Anyway, some of them appear different times due to the fact that the same TF can bind different positions along the promoter sequence. If we look in more detail, we can affirm that the TF with more possibilities to bind our promoter sequences is Sp1 [T00759] because it appears multiple times in the sequence, showing that it can bind different places along the promoter sequence. An important aspect to pay attention to is that in this table the only TF which coincides with the TFs file is HIF-1 [T01609], which has a Re equally value of 0.07.
Next step we did was to increase a bit more the permitted Re equally value in order to determinate if another transcription factor could bind our problem sequence. Afterwards, we selected only the TFs which coincided with those present in the TFs matrix.
- The obtained results are resumed in the next table:
In this table we can see that, when increasing the Re equally value, YY1 [T00915] and RXR-alpha [T01345] TFs appear. YY1 [T00915] TF is rejected to bind our promoter sequence because it shows very high Re equally values. However, it is possible that also RXR-alpha [T01345] could bind, because it shows not very high Re equally values, about 0.27.
The obtained results from Perl program are the following:
After analyzing the table, we can see that the TF which matches with the PROMO results is HIF-1 [T01609]. It is important to say that the exact binding position of this TF to the promoter sequence is not the same for PROMO than for Perl. In the case of PROMO, it shows us that the binding position of HIF-1 [T01609] is from the 970th position to the 978th. However, for Perl, the binding position is from the 478th to the 486th. The fact that the exact positions do not match can be basically due to the fact that the given TF matrices and the ones used by PROMO are different.
We did the same selection process than in AML1, and we obtained the next results:
In the table we can see that only 3 TFs do appear, although all of them come out some times along the promoter sequence. If we look at it with more detail, we can affirm that the TF with more possibilities to bind our promoter sequence is GR-beta [T01920] because it appears multiple times in the sequence, indicating that it can bind different places along the promoter sequence. It is important to say that any of the TFs given in the file do appear. Because of that, we decided to increase a bit more the maximum Re equally value, but always conserving the 5% dissimilarity. Once the new values were obtained, we built a table just showing the obtained values from our TFs which matched with our TF matrix.
- The obtained results are resumed in the next table:
Here we observe that AR [T00040], YY1 [T00915] and RXR-alpha [T01345] TFs do appear. Nevertheless, we can see that their values are too high. Thus, we claim that none of these TFs can bind our promoter sequence.
- The obtained results from Perl program are the following:
After analyzing the table, we can see that any TF fits with the obtained TFs in PROMO. At first, it should be considered that NF-kappaB [T00590] would be a candidate to be a TF binding our promoter sequence, because its p-value is very low (0.01). However, this TF does not appear in the PROMO program. One of the reasons could be that we have used a 5% dissimilarity. So, if we increase this value to 15%, for example, it could be possible that some of the TF in the matrix would appear.
We did the same selection process that in the previous cases and we obtained the next result:
In the table we can see that only 5 TFs come into view, appearing each of them some times along the sequence. Looking in more detail, we can affirm that the TF with more possibilities to bind our promoter sequence is GR-alpha [T00337], because it appears many times in the sequence, indicating that it can bind different places along the promoter sequence. It is important to mention that it does not appear any of the TF attached in the given file, so we decided to increase a bit more the maximum Re equally, but still conserving the 5% dissimilarity. Once the new values were obtained, we performed a table showing only the obtained values for the different TFs which fitted our TF matrix.
- The obtained results are resumed in the next table:
Now we can see that AR [T00040], YY1 [T00915] and RXR-alpha [T01345] do appear. However, we observe that the values are too high, so we reject the possibility that any of these TFs could bind our promoter sequence.
- The obtained results from Perl program are the following:
After analyzing the table we can see that any TF fits with the set TFs with PROMO. At first, it was considered that NF-AT1 [T00550] could be a candidate to be a TF which binded our promoter region, because its p-value was very low (0.06). Nonetheless, this TF did not appear in PROMO. One of the reasons could be that we had used 5% dissimilarity. So, if we increased this value until, for example, 15% it could be possible that one of the TFs in the matrix would appear.
RUNX1, AML1, CBFA2: Runt-related transcription factor 1
- The Gene Ontology term associations:
Nucleus [GO:0005634]
ATP binding [GO:0005524]
Protein binding [GO:0005515]
Transcription factor activity [GO:0003700]
Transcriptional activator activity [GO:0016563]
Hemopoiesis [GO:0030097]
Multicellular organismal development [GO:0007275]
Positive regulation of angiogenesis [GO:0045766]
Positive regulation of granulocyte differentiation [GO:0030854]
positive regulation of transcription from RNA polymerase II promoter [GO:0045944]
regulation of transcription, DNA-dependent [GO:0006355]
The AML1 gene found on chromosome 21 band q22, encodes a transcription factor (activator) for various haematopoietic-specific genes. It binds to the core site 5' PyGPyGGTPy 3' of a number of promotors and enhancers, as in GM-CSF (granulocyte-macrophage colony stimulating factor), CSF1R (colony stimulating factor 1 receptor), TCRb sites (T cell antigen receptors), and myeloid myeloperoxidase.
AML1 contains an N-terminal DNA-binding domain that is homologous to the Drosophila pair-rule gene runt. Normal AML1 protein (also known as CBFA2) is the DNA-binding subunit of the enhancer core-binding factor (CBF) and functions as a heterodimer with the non-DNA-binding subunit CBF that enhances AML1's DNA-binding affinity.
The heterodimeric Aml1 alpha2/beta transcription factor complex binds core enhancer sequences in the regulatory regions of several genes that are important to hematopoietic cell differentiation. Chromosomal aberrations in specific subsets of leukemia target the genes encoding either subunit of the complex to create transdominant chimeric oncoproteins.
A chromosomal aberration involving RUNX1/AML1 is a cause of the therapy-related myelodysplastic syndrome (T-MDS). It consists on a translocation t(3;21)(q26;q22) with EAP, MDS1 or EVI1. Moreover, a chromosomal aberration involving RUNX1/AML1 is a cause of chronic myelogenous leukemia (CML), which consists on t(3;21)(q26;q22) with EAP, MDS1 or EVI1.
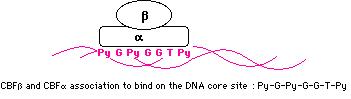
MDS1: Myelodysplasia syndrome 1 protein
The Gene Ontology term associations:
- Transcription factor activity [GO:0003700]
MDS1 is a separate gene which is located upstream of EVI1 on chromosome 3. It codifies for a transcription factor of unknown function. The fusion protein MDS1/EVI1 is also expressed normally through intergenic splicing.
EVI1: Ecotropic virus integration site 1 protein homolog
The Gene Ontology term associations:
- Nucleus [GO:0005634]
- Molecular function [GO:0003674]
- Biological process [GO:0008150]
EVI1 was originally identified as a frequent site of retroviral integration in murine myeloid tumour cells. It encodes a DNA binding protein on chromosome 3. Evi1 protein contains two domains of seven and three sets of repeats of the zinc finger motif, a repression domain between the two sets of zinc fingers and an acidic domain at the C-terminal end.
Because of the spatial and temporal restricted pattern of expression of EVI1, it is suggested that this gene plays an important role in mouse development and could be involved in organogenesis, cell migration, cell growth and differentiation.
EVI1 is normally expressed in non-hematopoietic tissues but abnormally expressed in haematopoietic cells. The inappropriate expression of the EVI1 in haematopoietic cells alters the terminal differentiation of bone marrow progenitor cells to granulocytes, erythrocytes and megakaryocytes. These EVI1 chromosomal disarrangements lead to Acute Myelogenous Leukemia (AML), Myelodysplastic Syndrome (MDS) and Chronic Myelogenous Leukemia (CML).
AME fusion protein
The AML1/MDS1/EVI1 chimeric gene is generated by the t(3;21)(q26;q22) translocation and plays an important role in the progression of haematopoietic stem cell malignancies such as chronic myelocytic leukemia and myelodysplastic syndrome.
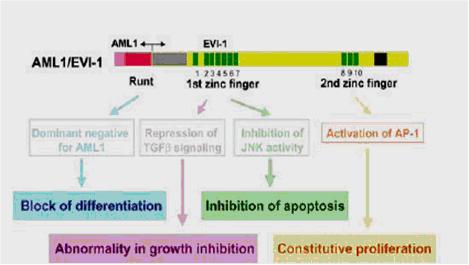
For a better understanding of the AME function we display the upper figure, which describes the AML1/EVI1 fusion protein. MDS1 codifies for a transcription factor with no important function or domains, which is located between AML1 and EVI1 in the fusion protein. So, as the relevant domains are found in AML1 and EVI1, this figure is perfectly useful for our willing.
In the AME fusion protein, an N-terminal half of AML1 including a runt homology domain is fused to the entire zinc-finger EVI1 protein.
There are 4 mechanisms identified in AML1/EVI1 fusion protein that possibly lead into malignant transformation of haematopoietic stem cells:
1) AML1/EVI1 exerts dominant-negative effects over AML1-induced transcriptional activation. Although target genes repressed by AML1/EVI1 are still unknown, binding competition to a specific DNA sequence and histone deacetylase recruitment through a co-repressor CtBP (C-terminal Binding Protein) in EVI1 part are plausible mechanisms for the dominant-negative effects.
2) AML1/EVI1 interferes with TGFb signalling and antagonizes the growth-inhibitory effects of TGFb. The first zinc-finger domain of EVI1 associates with Smad3, a TGFb signal transducer, and represses its transcriptional activity by recruiting histone deacetylase through CtBP that interacts with EVI1.
3) AML1/EVI1 blocks JNK activity and prevents stress-induced apoptosis. AML1/EVI1 associates with JNK through the first zinc-finger domain of EVI1 and disturbs the association between JNK and its substrates.
4) AML1/EVI1 enhances AP-1 activity by activating the c-Fos promoter depending on the second zinc-finger domain of EVI1 and promotes cell proliferation.
All these functions cooperatively contribute to the malignant transformation of the haematopoietic stem cells by AML1/EVI1.
back to the beginning
Our Bioinformatics project consisted in the full computational study of a protein given. It was a three-protein fusion that was composed by Evi1 protein, Mds1 protein and Aml1 protein. AML1 gene is located at chromosome 21, while EVI1 and MDS1 genes are both located at chromosome 3. First and foremost, we would like to start thanking our respective families and friends. We feel so grateful because of their unconditional comprehension and support. Thanks for understanding not only our full-time living at university in the last days but also our stress, bad mood and nerves when arriving at home.
Having finished this project, we have learned that chromosomal abnormalities are common genetic bases of leukemias. Many of these cytogenetic changes result in the creation of fusion proteins that contain transcription factors. The study of the role of these fusion transcription factors in leukemogenesis is important for understanding the molecular mechanism of leukemias.
Human t(3;21)(q26;q22) is a reciprocal translocation between chromosomes 3 (at band q26) and chromosome 21 (at band q22). It has been found as a secondary mutation in some patients with chronic myelogenous leukemia during blast phase (CML-BC), therapy-related myelodysplasia/acute myelogenous leukemia (t-MDS/t-AML) and in certain de novo acute myelogenous leukemia (AML). The t(3;21) translocation can result in the fusion of AML1 gene on chromosome 21 to several genes on chromosome 3, being the case of MDS1 and EVI1 in our protein 7. The fusion protein resulted from AML1-MDS1-EVI1 encodes a chimeric transcription factor of 200 kDa approximately. The AME fusion protein retains the DNA-binding domains of AML1 and EVI1. Thus, this chimerical transcription factor shows dual functions of AML1 and EVI1: differentiation block (due to Runt) and stimulation of proliferation (from the Zn fingers).
AML1, MDS1 and EVI1 are transcribed from telomere to centromere (reverse direction). When the t(3;21)(q26;q22) translocation occurs, the 3 genes are transcribed one after the other following this pattern: AML1-MDS1-EVI1. Therefore, MDS1 and EVI1 genes become regulated by AML1 promoter. Consequently, the expression of the AME fusion protein will be observed in the tissues and cell types where AML1 is regularly expressed. As we have explained in the gene expression analysis, highest Aml1 levels are found in thymus, bone marrow and peripheral blood. The fact that AML1 plays a crucial role in definitive haematopoiesis and blood vessel development explains that AME protein leads to acute myelogenous leukemia.
Nonetheless, the role of the AML1-MDS1-EVI1 fusion gene in leukemogenesis is still unknown. This is due to the fact that the development of leukemia is a complex process that may involve both the effect of the fusion genes in correct target cells and interactions of the target cells with the rest of the in vivo environment. Further biological studies and functional analyses of the AML1-MDS1-EVI1 fusion protein should provide useful insights into the mechanism of oncogenesis mediated by the alteration of transcriptional control.
back to the beginning
ACKNOWLEDGEMENTS
Thank you to all of our classmates who have kindly helped us with Perl programming and with any practical advice about the project. Also, thanks for your enthusiasm and encouragement during the eternal hours in front of the computer.
Last but not least, we would like to thank our B-group supervisors Pablo Martínez and Simon Furney for all their guidance and solved doubts. Moreover, we must express our extreme gratitude to Hagen Tilgner and Charles Chapple, the C-group supervisors, for the extra-hours they spent helping us with the arduous Perl program.
To everybody who has collaborated in making the realization of this Bioinformatics project a bit more bearable -no matter the way- thank you very much!

- Web sites:
http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3 [March 2007]
http://atlasgeneticsoncology.org [March 2007]
http://www.ensembl.org [March 2007]
http://www.geneontology.org [March 2007]
http://genome.ucsc.edu [March 2007]
http://www1.imim.es/courses/BioinformaticaUPF [March 2007]
http://www.ncbi.nlm.nih.gov [March 2007]
http://www.ebi.ac.uk/clustalw/ [March 2007]
- Articles:
Cuenco GM, Nucifora G, Ren R. Human AML1/MDS1/EVI1 fusion protein induces an acute myelogenous leukemia (AML) in mice: a model for human AML. Proc Natl Acad Sci USA. 15;97(4): 1760 - 5 (2000).
Fears S, Mathieu C, Zeleznik-Le N, Huang S, Rowley JD, Nucifora G. Intergenic splicing of MDS1 and EVI1 occurs in normal tissues as well as in myeloid leukemia and produces a new member of the PR domain family. Proc Natl Acad Sci USA. 20;93(4): 1642 - 7 (1996).
Ghozi MC, Bernstein Y, Negreanu V, Levanon D, Groner Y. Expression of the Human Acute Myeloid Leukemia Gene AML1 is Regulated by Two Promoter Region.
Proceedings of the National Academy of Sciences of the United States of America 93(5): 1935 - 1940 (1996).
Lacaud G, Gore L, Kennedy M, Kouskoff V, Kingsley P, Hogan C, Carlsson L, Speck N, Palis J, Keller G. Runx1 is essential for hematopoietic commitment at the hemangioblast stage of development in vitro. Blood 100(2): 458 - 466 (2002).
Liu XP, Zhang MR, Dai Y, Zhang L, Li R, Hao YS, Xiao ZJ. Study of genes involved in chronic myeloid leukemia with t (3; 21) (q26; q22) in blastic crisis. Zhonghua Xue Ye Xue Za Zhi 27(5): 310 - 3 (2006).
Mitani K. Molecular mechanisms of leukemogenesis by AML1/EVI-1.Oncogene 23, 4263 - 4269 (2004).
Mitani K, Ogawa S, Tanaka T, Miyoshi H, Kurokawa M, Mano H, Yazaki Y, Ohki M, Hirai H. Generation of the AML1-EVI1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukaemia. The EMBO Journal 13(3): 504 - 510 (1994).
Vinatzer U, Mannhalter C, Mitterbauer M, Gruener H, Greinix H, Schmidt HH, Fonatsch C, Wieser R. Quantitative Comparison of the Expression of EVI1 and Its Presumptive Antagonist, MDS1/EVI1, in Patients with Myeloid Leukemia. Genes, Chromosomes & Cancer 36: 80 - 89 (2003).
back to the beginning