Supervision: Eduardo Eyras, (eduardo.eyras at upf.edu) in collaboration with Josep Vilardell (josep.vilardell at crg.es)
RNA Splicing
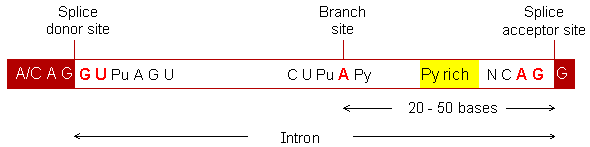
RNA splicing is a process that removes introns and joins exons in a primary transcript (pre-mRNA). Introns contain signals that are recognised by protein complexes (splicing factors) and produce the splicing. This is a typical intron in vertebrates:
Most introns start from the sequence GU and end with the sequence AG (in the 5' to 3' direction). They are referred to as the splice donor and splice acceptor site, respectively. However, the sequences at the two sites are not sufficient to signal the presence of an intron. Another important sequence is called the branch site located 20 - 50 bases upstream of the acceptor site. The consensus sequence of the branch site for vertebrates is "CU(A/G)A(C/U)", where A is conserved in all genes. In over 60% of cases, the exon sequence is (A/C)AG at the donor site, and G at the acceptor site.
The detailed splicing mechanism is quite complex. In short, it involves five small nuclear RNAs (snRNAs) and their associated proteins. These ribonucleoproteins (RNPs) form a large complex, called spliceosome. Then, after a two-step enzymatic reaction, the intron is removed and two neighboring exons are joined togethee. The branch point A residue plays a critical role in the enzymatic reaction:
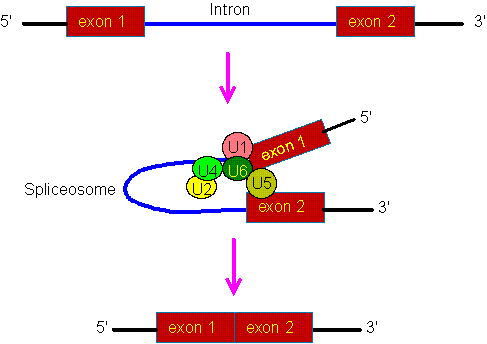
The figure shows a schematic drawing for the formation of the spliceosome during RNA splicing. U1, U2, U4, U5 and U6 denote snRNAs and their associated proteins. The U3 snRNA is not involved in the RNA splicing, but is involved in the processing of pre-rRNA.
The sequence signals defining introns in Yeast species are much more conserved than in vertebrate species:

The majority of Yeast introns have a donor splice sites corresponding to the hexamer GTATGT, and a donor site of mostly TAG or CAG but never GAG. Additionally, most of the introns have the branch site sequence TACTAAC.
Real introns that depart significantly from the consensus may be regulated by additional signals. That is,
other signals in the intron that promote the recognition and splicing of the intron.
These mecahnism may occur by the binding for molecules on intron signals.
The objective of this project is to search for signals conserved
in the introns for the cases where the splice sites and branch site sequences are different from the consensus.
We want to find out whether in cases where these sites depart from the consensus, there
are regulatory signals that are responsible for these splice-sites still being recognised.
Data set: this is a file with the multiple alignments of 163 introns of Saccharomyces cerevisae to other 3 Saccharomyces species: S. bayanus, S. mikatae and S. paradoxus.
In these alignments there are 126 cases for which the donor splice site is "GTATGT" in S. cerevisae, and 143 cases for which the branch site is "TACTAAC" in S. cerevisae. These sequence signals are not necessarily conserved in all other Yeas species in the alignments.
The file of the Data set contains multiple alignments like this one:
Sc_YAL001C GTATGTTCATGTCTCATTCTCCTTTTCGGCTCCGTTTAGGTGATAAACGTACTATATTGTGAAAGATTATTTACTAACGACACATTGAAG
Sb_YAL001C GTATGTATACGCTTCATCCCC-GCTCTAGCTTCCCTCATTCGAGAAGCATGCTGTGT-GCAAGAAAGCCTTTACTAACTATTTGTTAAAG
Sm_YAL001C GTATGTCTATGTCTCATTTTT-TTTGCAGTCTTTCTCTCGTGAGGAATGTTCTATTTTGTAAGAGATCTTTTACTAACTACTTACCAAAG
Sp_YAL001C GTATGTATATGCCTCATTCTTCTATTCCGTTCTTTTCAGGTGAGAAACGTGATATATTGT--AAGATTATTTACTAACGACTTATTAAAG
****** * * **** * * * ** * * * * * * * * ********* * ***
In this example, these lines represent the alignment of an intron from gene YAL001C in Saccharomyces cerevisae to the other 3 Saccharomyces species. The last line indicates the conserved positions with an asterisk.
you can read about an analysis of the sequence signals defining the introns in Yeast species.
We will assume that the alignments are correct, hence we assume that the signals that are conserved are also in the same position in the alignment. For instance, in the example above of YAL001C we can see there is a sequences TCAT conserved in the intron.
You will apply this second program to the two groups of multiple alignments. The aim is that you find the conserved signals (k=4,5 and 6) that appear in each group. Those signals that appear in the group of the introns with non-standard splicing signals but do not appear in the group of the introns with standard splicing signals are candidates for splicing promoter signals.
#!/usr/bin/perl -w
use strict;
my %hash;
my @species = ("Sc","Sb","Sm","Sp");
my $current_gene_id;
while(<>){
# we are reading a file with several multiple alignments like this one
#Sc_YAL001C GTATGTTCATGTCTCATTCTCCTTTTCGGCTCCGTTTAGGTGATAAACGTACTATATTGTGAAAGATTATTTACTAACGACACATTGAAG
#Sb_YAL001C GTATGTATACGCTTCATCCCC-GCTCTAGCTTCCCTCATTCGAGAAGCATGCTGTGT-GCAAGAAAGCCTTTACTAACTATTTGTTAAAG
#Sm_YAL001C GTATGTCTATGTCTCATTTTT-TTTGCAGTCTTTCTCTCGTGAGGAATGTTCTATTTTGTAAGAGATCTTTTACTAACTACTTACCAAAG
#Sp_YAL001C GTATGTATATGCCTCATTCTTCTATTCCGTTCTTTTCAGGTGAGAAACGTGATATATTGT--AAGATTATTTACTAACGACTTATTAAAG
# ****** * * **** * * * ** * * * * * * * * ********* * ***
#
chomp;
my ($id,$seq) = split;
next unless ($seq && $seq=~/[ACGT]+/);
$id =~/(S\w)_(\S+)/;
my $species = $1; # the yeast species
my $this_gene_id = $2; # the gene id that we are reading at this line
if (!$current_gene_id){
$current_gene_id = $this_gene_id;
}
# the gene id for this multiple alignment
if (!($current_gene_id eq $this_gene_id)){
# before turning to the next gene id, we can check the
# splicing signals and conservation for the four sequences:
for( my $i=0; $i< scalar(@species); $i++){
my $species = $species[$i];
print $species."\t".$current_gene_id."\t".$hash{$species}{$current_gene_id}."\n";
}
$current_gene_id = $this_gene_id;
}
# build a table with the information for this multiple alignment:
$hash{$species}{$current_gene_id} = $seq;
}
Try this example program and verify that it works.
The students will present the methods and results of their work in a web page. They should describe the methods they have used, include the programs developed for the analysis and present the obtained results for k=4,5 and 6. The second program developed should in principle be able to work for any value of k (it should have k as a variable within the program).
Database of Yeast sequenced genomes Genolevures.
Database of Saccharomyces cerevisae introns Ares lab
SGD: Sccharamyces Genome Database SGD