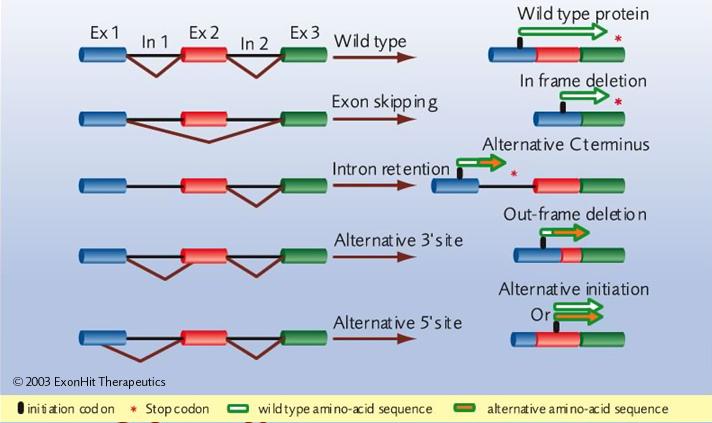
- Inclusió/exclusió d'exons ("exon skipping")
- Retenció d'introns
- Lloc de "splicing" 3' alternatiu
- Lloc de "splicing" 5' alternatiu
- Exons mútuament excluients
(no mostrat a la figura)
L' splicing alternatiu (SA) és un mecanisme pel qual el pre-mRNA pot donar lloc a
diferents combinacions d'exons, produďnt diferents mRNAs madurs. Així,
permet que es tradueixin diferents proteďnes del mateix gen i en modula
la funcionalitat.
Estudis computacionals realitzats estimen que un 40-60% dels gens humans pateixen SA.
L' splicing alternatiu és regulat per patrons espacio-temporals
específics durant el desenvolupament, i juga un paper important en la
regulació gčnica, per exemple en la determinació del sexe en
Drosophila, la resposta immunolňgica en humans o en processos
específics d'estadis del desenvolupament.
Hi ha cinc tipus principals de "splicing" alternatiu:
|
|
Sembla ser que la selecció natural esbiaixa la utilització de codons per millorar la síntesi proteïca en E.coli, S.Cerevisiae,C.elegans i A.thaliana. Aquestes espècies mostren correlacions positives entre el biaix de codons i els nivells d'expressió gènica. A més, els codons utilitzats preferentment, tendeixen a ser reconeguts per tRNAs abundants en E.Coli, bacillus subilis, llevadures, D.melanogaster i C.elegans. Aquests patrons suggereixen un paper de la selecció natural sobre els llocs sinònims i una diferència de fitness entre els codons sinònims (selecció traduccional)(1)
Així, podem relacionar el biaix de codons amb els nivells d'expressió gènica en les espècies esmentades.
En mamífers, la selecció traduccional als llocs sinònims no està massa clara. Es creu que les diferències en la utilització de codons sinònims en relació als nivells d'expressió són molt petites. Els estudis en mamíers es compliquen per l'heterogenicitat en la composició de bases dels genomes dels mamífers, ja que els cromosomes d'aquests semblen ser mosaïcs de llargs segments de DNA anomenats isocores (isochores) que tenen un contingut en GC distintiu. Així, per detectar la selecció sobre els llocs sinònims és important eliminar l'efecte de les isocores (1).
S'han proposat algunes evidències de selecció traduccional en mamífers, però estan poc fonamentades.
Amb aquest treball es pretén aprofundir una mica més sobre el biaix de codons sinònims en mamífers en relació amb el grau d'expressió, mitjançant un anàlisi computacional del biaix de codons en exons constitutius i alternatius de seqüències de mRNA de ratolí.
En els gens que pateixen 'splicing alternatiu', els codons dels exons que s'expressen constitutivament es tradueixen amb més freqüència que els dels exons alternatius. Seria d'esperar que la selecció traduccional actués amb més força sobre el biaix de codons i reduïs la divergència silent als exons constitutius que als alternatius. Així, els gens que codifiquen per proteïnes que pateixen 'splicing' alternatiu són una bona oportunitat per examinar exons que difereixen en els nivells d'expressió però que es troben a la mateixa isocora (pocs gens creuen els límits d'una isocora).
El biaix de codons és un tema emergent en genòmica evolutiva des de fa uns quants anys. Es disposa de multitud d'articles publicats relacionats amb aquest tema (a l'apartat Referències i Agraïments es mostren algunes revisions publicades recentment (2)(3)(4)(5)(6))
De totes maneres, en revisar la literatura científica, només s'ha trobat un treball previ del Institute of Molecular Evolutionary Genetics (Universitat de Pensilvània) que es plantegi la qüestió del biaix de codons diferencial en els exons alternatius i constitutius (1).
L'estudi analitza seqüències de gens que pateixen SA en humans i en Drosophila melanotgaster, amb l'objectiu d'estudiar la selecció traduccional. Els resultats mostren que els codons acabats en CG són més abundants en els exons constitutius que en els alternatius tant en Drosophila com en humans.
L'objectiu d'aquest projecte és estudiar l'ús de codons en els
esdeveniments d'exclusió d'exons, és a dir, comparar la distribució de
codons en els exons alternatius respecte als exons constitutius.
En concret, l'estudi es realitzarŕ a partir d'un arxiu de seqüčncies de mRNAs de ratolí que pateixen SA.
En el següent enllaç podeu observar l'arxiu: Seqüčncies d'exons de ratolí
Per tal d'obtenir l'arxiu només cal que cliqueu sobre el link amb el botó dret i seleccioneu "Save link as". Assegureu-vos de guardar tots els arxius a la mateixa carpeta (seria convenient que creessiu una carpeta només per l'ús d'aquest treball.
Si ho preferiu, podeu descarregar-vos el fitxer comprimit: mouse.skipping.seqs.gz
Per tal de descomprimir-lo useu la comanda gzip -d.
I aquí teniu explicat com llegir el fitxer: Instruccions per a llegir el fitxer
Per a realitzar l'estudi se seguiran els següents passos:
S'ha escrit un programa en llenguatge Perl (EXAL) i s'ha executat sobre el fitxer de seqüències ("mouse.skipping.seqs.txt") cridant-lo des del Shell amb la següent comanda:
$ exal.pl mouse.skipping.seqs.tx
Link amb el programa EXAL: EXAL
Explicació del programa:
La primera línia de programa assigna el nom de l'arxiu.txt a una variable i, posteriorment l'obre pel seu anàlisi.
L'estudi requereix un anàlisi línia a línia del fitxer de text. Així, per a cada línia s'haurà de fer el tractament que calgui i anar emmagatzemant les dades a mesura que es vagin llegint més línies..
S'assigna cada columna de la línia a una variable mitjançant una expressió regular que encaixa tot el que són caràcters separats per tabulacions. Això ens permetrà tractar les dades amb més comoditat. Algunes dades de de les què assignem a variables no s'utilitzaran posteriorment, però s'ha considerat una forma de visualitzar totes les dades que conté el fitxer.
A continuació es troba la base del nostre programa: comptar els codons que apareixen a cadascun dels tipus d'exons. Se separaran els codons en tres grups: els dels exons constitutius (exons 1 i 3), els dels exons alternatius (exó2) i els codons que queden partits entre dos exons degut a la fase de l'exó següent (despenjats).
En un primer pas s'agafa la part codificant de cada seqüència exònica, la qual cosa s'aconsegueix mitjançant la funció substr:
$exoX = substr ($seqX,$startreal,$endX - $startX + 1);
Per a la funció "substr" es defineixen els següents paràmetres:
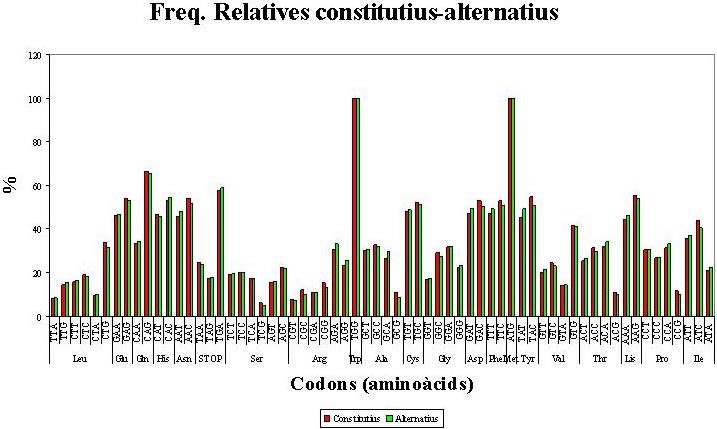
En les freqüències relatives d'ús de codons entre els exons constitutius i alternatius, representades gràficament a la Figura 1 , s'observen lleugeres variacions, però es manté sempre l'ordre de prioritat d'utilització dels codons sinònims.
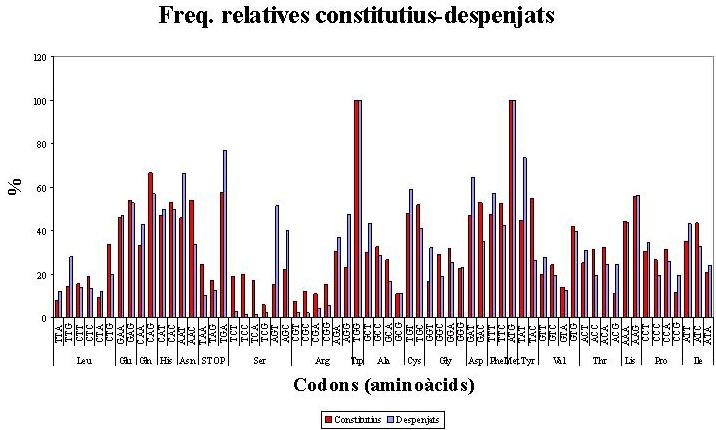
Les diferències de freqüència entre els codons constitutius i els despenjats (Figura 2) són més marcades. En aquest cas no sempre es manté l'ordre de prioritat en l'ús dels codons sinònims. Cal tenir en compte que estem comparant dos mostres de mida molt diferent. Aquesta comparació no té un sentit biològic des del punt de vista dels objectius del treball. S'ha considerat interessant representar les dades en una gràfica però no es tindran en compte per a les conclusions del treball.
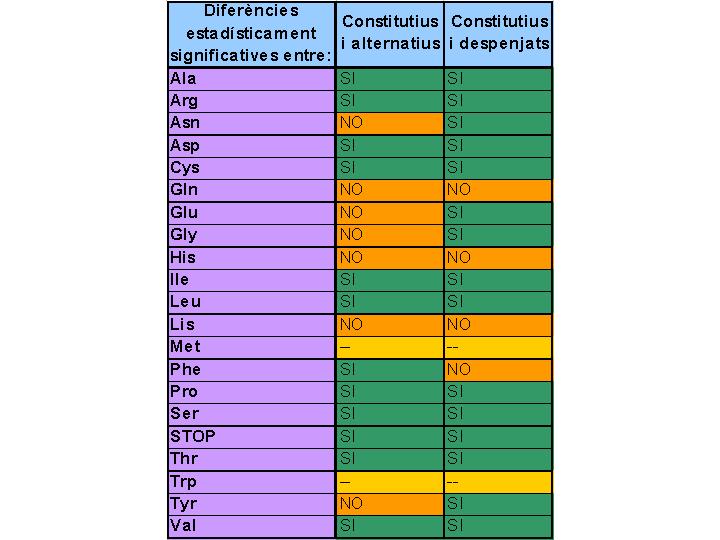
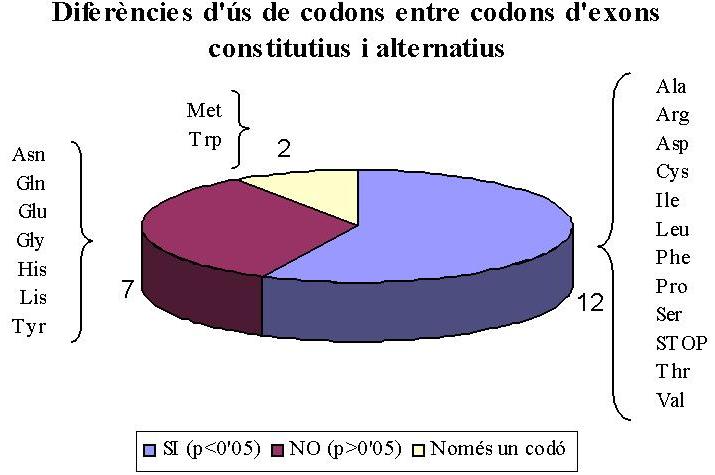
La significació de les diferències observades en les gràfiques anteriors es recull en la taula de la Figura 3, construïda a partir dels resultats del test de la chi-quadrat. La significació s'ha establert per a P < 0,05. Les mateixes dades es recullen en forma de diagrama de sectors a la Figura 4. S'han trobat diferències significatives en la utilització dels codons en 11 dels 18 aminoàcids amb codons sinònims (Ala, Arg, Asp, Cys, Ile, Leu, Phe, Pro, Ser, Thr, Val) i en els codons de STOP. Els aminoàcids Asn, Gln, Glu, Gly, His, Lys, Tyr, en canvi, no mostren diferències significatives. Els aminoàcids Met i Trp no tenen codons sinònims i, per tant, no poden mostrar diferències. S'ha observat una predominància d'aminoàcids hidrofílics en els que no mostren diferències (són tots hidrofílics excepte Gly i Tyr).