

Ana Igea Fernández, Iris Uribesalgo Micás
Les données à partir desquelles nous partîmes étaient des 60770 cDNAs de souris, à l'intérieur desquels certains étaient décris comme des codificateurs, et d'autres comme non codificateurs. Tous étaient théoriquement clonés en forward (référence article).
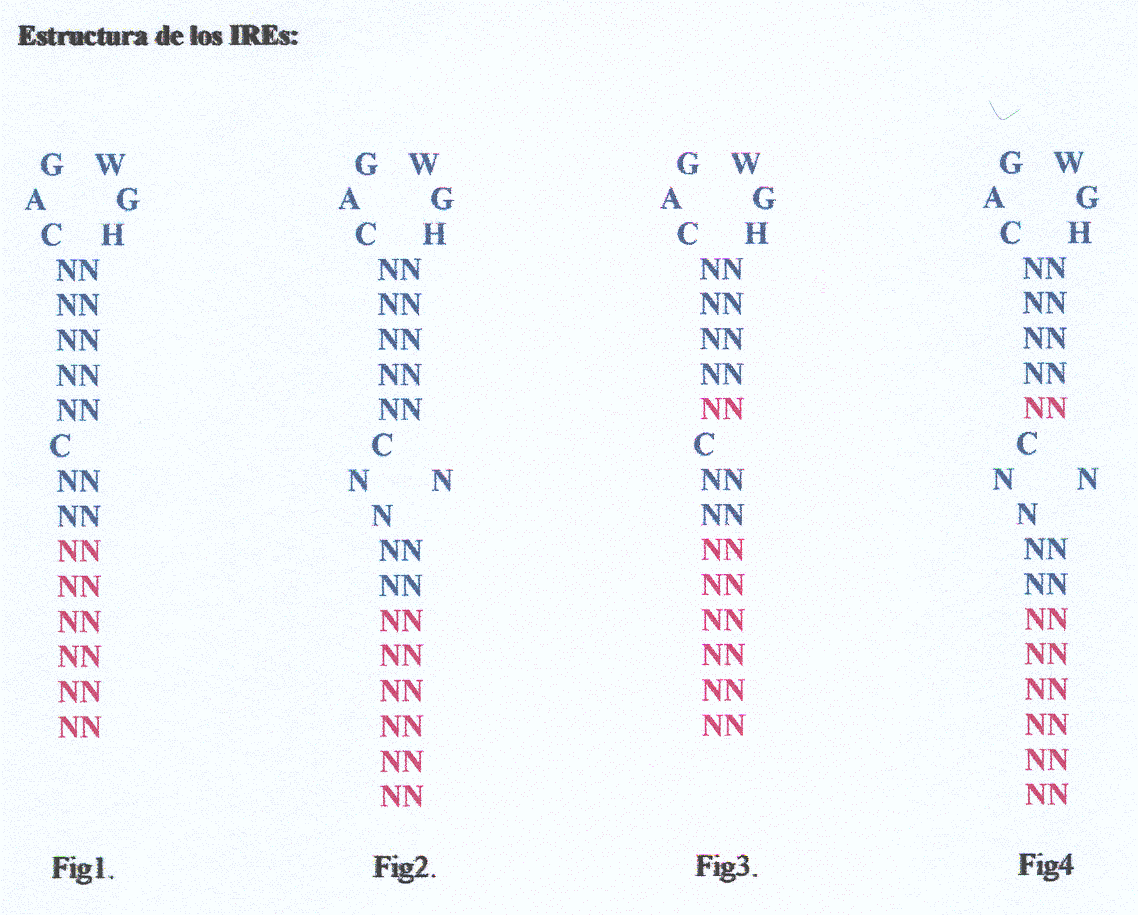
Pour commencer nos travaux, nous nécessitions créer dans un premier temps un modèle qui nous définisse les structures des IREs que nous étions intéressés de trouver. A partir de ces modèles d'IREs réalisées l'année passée et en nous basant aussi sur une bibliographie d'articles [1,2,3,4,5,6,7,8] , nous avons décider d'utiliser deux modèles de prédiction différents:
Nous avons décidé d'utiliser dans un premier temps deux modèles étant donné que, en fonction du type de protéïne, les modèles d'IREs diffèrent considérablement (modèles) . Par conséquent, nous avons pensé que, avec un modèle plus laxiste, nous pourrions inclure les IREs d'une grande majorit´ de protéïnes (si ce n'est toutes). Pour terminer la définition la structure de ce modèle, nous nous basons dans les données suivantes trouvées dans la bibliographie:
En utilisant deux modèles, nous nous sommes présenté la possibilité que le nouveau modˇèle plus laxiste fasse augmenter le nombre de résultats obtenus et, par conséquent, le nombre de faux positifs. Nous devions juger, au vu des premiers résultats de structures IRE possibles, de la pertinence du modèle laxiste i.e. s'il nous est utile ou si, au contraire, il inclus trop de structures non spécifiques.
Nous avons utilisé le programme Scan_for_matches pour balayer mes modèle sur les cDNAs de de la séquence.
Le premier dilemme était de savoir si nous devions balayer les 60770 cDNAs de souris ou simplement sur les cDNAs décrits comme codificateurs et, d'autres par si nous deviosn effectuer le balayage en "forward" ou en "reverse".
Dans un premier temps, nous avons décidé d'utiliser les 60770 cDNAs de souris pour ętre sűrs de ne trouver aucun IRE dans des régions non codifiées et vérifer la pertinence des résultats sur les régions que l'article de référence qualifiait comme non codifiées. Dans un deuxième temps, nous avons de balayer en forward ainsi quen reverse chacun des patrons et inscrire les résultats dans des archives distinctes pour une analyse ultérieure (IRESpatroreverse.txt, IRESpatroforward.txt, IRESpatrolaxereverse.txt, IRESpatrolaxeforward.txt):
Devant ces résultats, nous avons décidé de continuer de travailler sur les séquences obtenues à partir d'un modèle laxiste étant donné quil inclut un plus gran nombre de structures IREs possibles. La différence avec les séquences obtenues avec un modèle strict n'est pas tr&eagrave;s significative. Par contre, nous devrons ętre assez stricts lors de la validation thermodynamique (link al trabajo), et lorsque nous effectuerons la validation pas homologie avec le génome humain (Blast)
La décision suivante que nous avons pris a été de travailler à partir des séquences obtenues avec les résultats du modèle laxiste, uniquement avec les données obtenues en forward. Cette décision prise avec l'appui des résultats du groupe chargé de la prédiction de gènes (link al trabajo), ledit groupe a exécuté le programme "geneid" avec les 60770 cDNAs en déduisant que, męme q'il y avait quelques cDNAs en reverse, ceux-ci étaient peu nombreux et la majorité d'entre eux n'avaient pas une bonne ponctuation. En nous basant avec ces données, et męme si quelques cDNA en reverse avaient une bonne ponctuation, nous avons cru qu'il était plus pertinent e travailler úniquement en forward męme si nous pouvions perdre quelques informations du fait de la difficulté ajoutée qu'est de travailler en reverse face à la faible possibilité de trouver quelques structure IRE dans ces cDNAs.
Une fois tous les points cités précédemment mis au clair, nous avons travaillé avec le fichier des séquences obtenues en forward à partir du modèle laxiste qui contien une prédiction de 601 IREs issus de 594 cDNAs différents.
A ce moment précis, il a été nécessaire de transformer le fichier que nous avions en langage fold en lengage fasta afin qu'il puisse ętre validé avec d'autres problèmes qui nécessitent ledit langage. Pour ce faire, nous avons créé un programme en PERL RNAfold_fasta.pl, celui-ci a converti nos séquences en éliminant les espaces entre nucléotides et en changeant les Ts en Us (IRESpatrolaxeforward.fasta).
Pour valider nos résultats, nous avons réalisé un Blast des 601 séquences correspondant à de possibles IREs contre le génome humain étant donné que la majorité des IREs trouvés dans la souris devraient figurer dans le le génome humain (99% du génome est conservé et les IREs, par ailleurs, survivent au splicing et ont une fonction concréte).
"Blast" es une ressource que nous pouvons trouver sur el site ou que nous pouvons directement obtenir depuis UNIX. Nous avons décidé d'utiliser l'application depuis UNIX, pour cela, nous avons utilisé uen commande spécifique en plus de créer une variable pour nous économiser un bucle avec la finalité que la ressource Blast parcoure tous els cromosomes [4].
Avant d'utiliser Blast nous devoions décider:
Une foit nous avions les donées assurées nous avons fait courrir le Blast de les 601 séquences de possibles IREs sur la base de donées dű génome humaine masqué, mais il n'etâit possible le finaliser parce que l'ordinateur persy n'avait la puissance nécessaire.Ce pour ça que nous avons decidé courrir les Blast avec autre ordinateur pour utiliser cette programme qui s'appele Blastmachine est à plus puissance avec base de donnés grands comme la nôtre [5].
Ce qui paraissait une solution n'avait resultée.Le Blastmachine avit aussi des problemes. La prochaine solution ętait faire le Blast cromosome par cromosome avec une valeur de 0.1 d'Expected value [6], mais nous avons le męme problème de puissance de l'ordinateur.
La dernière solution ętait réduir la base de donnés en utilisant les ESTs humaines et non tout le génome masqué. On peut fait ça parce que les IREs sont transcrits et peuvent résistir a le "splicing".Aussi nous éliminons des intrones et régions intergenetiques qui peuvent donner une grand nombre des faux possitives. Cette fois le Blast fonctionait mais les (résultats) obtenus etaient peu significatives,doit à la grandeur de la base de donées. Une sólution qui nous aparait alors etait la possibilité de courrir un patron plus laxiste sur une base de donnés de cDNAs humaines crée avec une base de ESTs humaines disponibles dans la web pour pouvoir utiliser plus tard des séquences que nous obtendrions comme base de donées pour faire un nouveau Blast avec des 601 séquences obtenus avec le męme patron de les 60770 cDNAs de souris [7].
C'est ainsi comme nous atteindons travailler seulment avec des régions codifiants (évitant les intrones et régions intergenetiques) et nous obtenons une réduction de la base de donées pour obtener des hits plus spécifiques et moins faux positives. Ce Blast ętait aussi impossible de réaliser,
Nous avons creé un patron lequel nous a donée des séquences qui contenaient genes avec des structures secondaires dans le 3' et le 5' UTR qui pouvaien correspondre a des IREs réals.
Cette prediction s'avait realisé sur les 60770 cDNAS de souris et les résultats ętaient verifiés pour hómologie avec le Blast contre ESTs humaines. Les résultats obtenus aprés tout ce procčs n'ętaient trop significatif comme nous avons dit anteriorment. Vous pouvez consultez ces réslultats dans le suivant link.
2-03-2003 E-mail: anuky4@hotmai.com ; yurcelay@hotmail.com
Avec celui-ci décrit en bibliographie [1] , comme le montre notre shéma, nous espérons trouver cinq appariements exacts dans la "stem" supérieur et deux à huit appariements dans la "stem" inférieure. De plus, nous acceptons les appariements A-T et G-C ainsi que les G-T étant donné quil a été observé que, męme si ces derniers ne soient pas des appariements classiques, ils se retrouvent dans un grands nombre de structures secondaires.
Ce modàle, que nous avons définis à partir de données écrites dans la bibliographie, difère de la précédente dans le fait que quatre ou cinq appariements sont permis dans la "steam" supérieure.
2. Exécution du programme "Scan_for_matches"
3.Traduction des résultats en lengage FASTA
4. BLAST contre génome humain
5. Résultat
