1.INTRODUCCIÓN
La teoría de Darwin esta totalmente aceptada: 'Todas las especies actuales y las extinguidas provienen de un origen ancestral común, que a lo largo de la evolución se han ido diferenciando unas de otras, formando nuevas especies'.
Esta teoría proviene exclusivamente de la observación del fenotipo de las especies, pero entenderla significa comprender la evolución de los genomas que se ha dado paralelamente.
Hace poco más de veinte años que se conoce la presencia de intrones en los genomas eucariotas. Su ausencia en los genomas procariotas es una primera evidencia de evolución molecular.
Dos hipótesis contrarias explican la existencia de los intrones:
Según la teoría Introns early, los intrones han existido siempre funcionando como espaciadores de antiguos minigenes monoexónicos, que se perdieron en Bacteria y Archaea y se mantienen en Eucarya.
En cambio la teoría de Introns late considera que los genes iniciales no tenían intrones, eran exones puros, y que la producción de intrones se dio como resultado de inserciones durante la aparición de células eucariotas.
Ninguna de las dos hipótesis se ha considerado más válida que la otra, debido a que hay evidencias que favorecen una o la otra.
Han sido descritos diferentes mecanismos de evolución genómica, que intentan explicar cómo los genomas adquieren complejidad a lo largo de la evolución.
La teoría del exon shuffling afirma que la duplicación, inserción y deleción de exones son tres tipos de procesos que intervienen en la evolución de nuestro genoma, creando nuevos genes.
Se entiende por duplicación, la duplicación de uno o más exones dentro de un mismo gen, de forma que el gen se hace más largo tras darse este proceso. Se entiende por inserción, el intercambio de uno o más exones entre diferentes genes, de forma que las proteínas codificadas por estos genes pueden adquirir nuevos dominios funcionales o estructurales tras darse este proceso, y finalmente se entiende por deleción la pérdida de uno o más exones de un gen, de forma que dicho gen puede perder función tras darse este proceso.
El resultado del shuffling son nuevas construcciones de exones en un gen.
La teoría entiende los exones como dominios estructurales que pueden o no venir acompañados de función, y explica como las proteínas pueden ganar nuevas funciones mediante la inserción de nuevos exones en un gen.
La 'exonización y pseudoexonización' hacen referencia a la creación o eliminación de �splicing sites� de forma que una secuencia intrónica pase a formar parte de una región exónica o viceversa. El resultado es la ganancia o pérdida de una secuencia codificante que puede dar lugar a una ganancia o pérdida de función.
Tras darse estos procesos las proteínas van ganando dominios funcionales, que pueden mantenerse o desaparecer a la largo del tiempo, según tengan una función más o menos conservada.
Según la teoría de introns late, los intrones pueden insertarse en medio de una región codificante del genoma a lo largo de la evolución, de forma que este intrón estará truncando la información que contenía dicha región.
Puede que esta información se pierda o que la selección purificadora (según la Teoría Neutralista de la evolución) no acepte esta pérdida.
Una posibilidad para recuperar esta información es juntar la información que ha quedado separada por la reciente inserción (intrón), es decir, que la información truncada se convierta en dos exones del gen.
Como resultado de este proceso, el dominio proteico codificado en esta región, estará separado en dos exones diferentes.
Las proteínas humanas están formadas por diferentes dominios proteicos, algunos de ellos muy conservados a lo largo de la evolución.
Estos dominios están codificados en regiones exónicas de los genes. Pero ...�Siempre se localizan dentro de un solo exón?
|
VOLVER A LA TABLA DE CONTENIDOS
|
2.OBJETIVOS
Para resolver el problema planteado abordamos los siguientes objetivos:
- Estudiar la localización de 433 patrones en unas 9.000 secuencias de proteínas, anotando si su localización es intra o interexónica.
- Encontrar aquellos patrones de longitud considerable que estan ubicados en
una región intraexónica con una frecuencia mayor que la esperada por azar
(patrones significativos o patrones con motivo de estudio)
- Comprobar si hay alguna relación entre las funciones de los patrones
considerados significativos.
- �Existe alguna razón evolutiva que explique el sesgo observado en estos patrones?
|
VOLVER A LA TABLA DE CONTENIDOS
|
3.MATERIALES Y MÉTODOS
Los archivos que hemos usado para lograr los objetivos de nuestro proyecto han sido:
El archivo refseq.aa.fa es subconjunto de refseq,
contiene la secuencia aminoacídica de más de 9000 proteínas
que están bien anotadas, en formato fasta:
>NM_013371
MKLQCVSLWLLGTILILCSVDNHGLRRCLISTDMHHIEESFQEIKRAIQAKDTFPNVTIL
STLETLQIIKPLDVCCVTKNLLAFYVDRVFKDHQEPNPKILRKISSIANSFLYMQKTLRQ
CQEQRQCHCRQEATNATRVIHDNYDQLEVHAAAIKSLGELDVFLAWINKNHEVMFSA*
El archivo de prosite que se encuentra en ftp://us.expasy.org/databases/prosite/release/prosite.dat,
contiene una lista de patrones protéicos. Para cada patrón
se encuentran los siguientes campos:
//
ID ASN_GLYCOSYLATION; PATTERN.
AC PS00001
DT APR-1990 (CREATED); APR-1990 (DATA UPDATE); APR-1990 (INFO UPDATE).
DE N-glycosylation site
PA N-\{P\}-[ST]-\{P\}
CC /TAXO-RANGE=??E?V
CC /SITE=1,carbohidrate
CC /SKIP-FLAG=TRUE;
DO PDOC00001;
Para poder usar los datos de este archivo en nuestro programa tuvimos que
hacer las siguientes modificaciones:
-De todos los datos en prosite.dat
sólo tomamos los campos AC y PA, es decir el identificador del patrón
y la secuencia del patrón, con la siguiente instrucción de Shell redireccionándolo al archivo prosite.tbl.
egrep"^AC|^PA" prosite.dat > prosite.tbl
-Formato de prosite.tbl: identificador del patrón seguido de la expresión regular
PS00001 N[^P][ST][^P]
PS00002 SG.G
PS00004 [RK][RK].[ST]
PS00005 [ST].[RK]
PS00006 [ST]..[DE]
Posteriormente ejecutamos un programa para poner los patrones en expresiones regulares para Perl,
y lo almacenamos en el archivio prosite.tbl pero con el mismo formato que
prosite.tab, archivo que contiene las posiciones donde comienzan
y terminan los exones de todas las proteínas de refseq en formato
tabular.
El primer elemento de la fila es el identificador de la secuencia;
el segundo elemento, separado por un espacio, es el identificador, un punto
y el número de exón; el tercer elemento es la primera posición
del exón y el marco de lectura nucleotídico separado por
un punto; el último elemento es la posición final del
exón y el marco de lectura nucleotídico separado por un punto
NM_000016 NM_000016.1 1.1 10.3
NM_000016 NM_000016.2 11.1 40.1
NM_000016 NM_000016.3 40.2 72.3
NM_000016 NM_000016.4 73.1 96.1
PATTERNmatching es el programa en lenguaje Perl sobre el que se basa nuestro proyecto,
su función es buscar patrones proteicos en secuencias e informarnos
sobre su ubicación intraexónica o interexónica.
El programa se divide básicamente en 3 partes:
- OBTENCIÓN DE ARCHIVOS.
En un principio, el algoritmo se encarga de obtener de los diferentes
archivos antes mencionados, la información necesaria para el proceso.
Para la ejecución del programa es necesario introducir previamente
el nombre de estos archivos en SHELL:
./patternmatching.pl <archivo de secuencias> <archivo de patrones><archivo de exones>
El programa verificará que se le hayan introducido
estos archivos y, en el caso de no ser así, nos avisará con
un mensaje de error. A continuación, se crean 3 filehandles que
permitirán la entrada de datos al programa. Además crearemos
otro filehandle para crear un archivo de salida (outfile.txt)que recopila
información que se obtiene a lo largo del proceso.
- PREPARACIÓN DE LA INFORMACIÓN.
En esta parte crearemos las diferentes matrices que se procesarán
a lo largo del algoritmo: matriz de secuencias, matriz de patrones y matriz
de exones.
- Matriz de secuencias: a partir de un archivo donde se almacenan
las secuencias en formato fasta, construimos una matriz en la cual el primer
elemento de cada vector hace referencia al ID de la secuencia (NM_xxxxxx)
y el segundo contiene la secuencia de aminoácidos. De esta matriz
obtenemos las secuencias sobre las cuales queremos encontrar los diferentes
patrones.
- Matriz de patrones: al igual que en la anterior matriz, transformamos
la información de un archivo de patrones obtenido de la base de
datos de prosite, en una matriz donde el primer elemento de cada vector
contiene los ID de los patrones (PSxxxxx) y el segundo la expresión
regular que definirá a cada patrón. De esta matriz obtenemos
los patrones que queremos buscar sobre las diferentes secuencias.
- Matriz de exones: el archivo de exones lo transformamos en una matriz que contiene
información sobre ID de proteína, número de exón,
posición inicial de exón y posición final de exón
de proteínas conocidas (Refseq). Esta es la matriz de consulta de la cual extraemos información acerca de la
distribución exónica de las diferentes proteínas,
es como una base de datos en la cual a partir del ID de la proteína,
obtenemos para cada exón que constituirá la proteína,
su posición inicial y final.
- Esta parte del programa consiste en
leer los archivos abiertos y extraer la información que contengan
transformándola en estas 3 matrices.
- CUERPO DEL PROGRAMA.
En esta parte del programa procesamos toda la información
que hasta ahora hemos obtenido. Consiste en buscar cada uno de los patrones
en cada una de las diferentes proteínas que constituye la matriz
de secuencias, para ello recorremos las matrices mediante varias composiciones
iterativas a la vez que aplicamos una función (match). Esta función
se encarga de "lanzar" la expresión regular que reconoce patrones
sobre cada una de las secuencias, y cada vez que encuentra una coincidencia
nos devuelve 3 valores: posición inicial del match, longitud del
match y secuencia de aminoácidos del match. Finalmente, la información
obtenida se almacena en un archivo de salida de nombre <OUTFILE.TXT> que
contiene información sobre cada uno de los patrones encontrados.
Para cada patrón obtendremos:
PATTERN
PSxxxxxx
Id. SEQUENCE NMxxxxxx
Total MATCHES x
MATCHx Posición inicial(longitud) ...secuencia aminoacídica...
Con esta información construiremos una nueva matriz (matriz de matches)
que nos servirá para continuar con el algoritmo. Esta matriz contiene
información acerca del ID del patrón, ID de la proteína
y posición inicial y final del patrón encontrado. Finalmente,
comparamos la matriz de matches obtenida con la matriz de exones para determinar,
a partir de las posiciones de los patrones encontrados y de los exones,
si los patrones reconocidos se ubican en el interior de un único
exón (intraexónico) o se extiende a lo largo de más
de un exón (interexónico).
Para ello, recorremos la matriz de matches hasta que coincidan los
ID, es decir, hasta que encontremos el ID de la proteína en el listado
de ID de proteínas con información sobre las posiciones exónicas.
Una vez recopilamos información sobre las posiciones inicial y final
de los exones solo basta comprobar, mediante una composición condicional,
que la posiciones inicial y final de los patrones encontrados caen o no
dentro de un exón.
Esta información aparecerá resumida por pantalla en
formato tabulado:
ID patrón, Longitud, coincidencias intraexónicas, porcentaje de intraexónicas,coincidencias interexónicas, porcentaje de interexónicas y total de patrones encontrados.
|
VOLVER A LA TABLA DE CONTENIDOS
|
4.RESULTADOS
Una vez ejecutado el programa PatternMatching, se obtiene una tabla de
resultados donde están representados los patrones encontrados una o más
veces dentro de cualquiera de las secuencias. A partir de esta tabla contruimos una tabla en Excel para poder analizar los datos estadísticamente.
Una primera observación consiste en ver qué patrones han hecho más matches.
Aunque se debe tener en mente que debido al tamaño del patron o a la
inespecificidad de la expresión regular que lo define algunos matches se pueden
producir por azar, la siguiente tabla muestra los patrones que dan lugar a la mayor parte de los matches totales:
Tabla de resultados 1
| ID DEL PATRóN |
LONGITUD DEL PATRóN |
FUNCIóN DEL PATRóN |
FRECUENCIA DE MATCHES |
| PS00006 |
4 |
Casein kinase phosphorilation site |
29.1 |
| PS00005 |
3 |
Protein kinase C phosphorilation site |
25.9 |
| PS00008 |
6 |
N-myristoylation site |
21.8 |
| PS00001 |
4 |
N-glicosilation site |
9.7 |
| PS00004 |
4 |
cAMP and cGMP-dependent protein kinase phosphorilation site |
3.57 |
| PS00007 |
7 |
Tyrosine kinase phosphorilation site |
2.86 |
| PS00009 |
4 |
Amidation site |
1.87 |
| PS00002 |
4 |
Glicosaminoglycan attachent site |
1.01 |
| PS00028 |
21 |
Zinc finger, C2H2 type, domain signature |
0.79 |
| PS00013 |
11 |
Prokaryotic membrane lipoprotein lipid attachment site |
0.43 |
Conclusiones...
A continuación realizamos, mediante una serie de análisis estadísticos,
una selección de los patrones significativos en función de su tama�o, para su
posterior análisis.
En primer lugar se ha relacionado la tabla de matches con la tabla de
descriptores de patrones extraida de Prosite, para poder relacionar patrón
y función. (tabla en Excel).
En segundo lugar, se ha obtenido una gráfica de distribución de longitudes
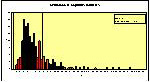
que representa la distribución de frecuencias de los patrones encontrados según su longitud.
Los patrones más encontrados son los de longitud 7, aunque la media se
situa sobre la longitud de 9. Se han clasificado los patrones según la
distribución de las longitudes de los patrones: aquellos que se desplazan
una desviación estándar por encima de la media (patrones de 17 aminoácidos
de longitud) se consideran patrones "largos" y aquellos patrones
inferiores a 17 aminoácidos de longitud se consideran "cortos".
Se han considerado significativos los patrones "largos", por lo que se proseguirá
el análisis a partir de estos.
Se han despreciado los patrones cortos porque tienen más probabilidad de
encontrarse dentro de un exón por azar, por lo que no son motivo de
análisis.
En tercer lugar se ha procedido a hacer un análisis individual de los datos
significativos.
Obteniendo una gráfica para cada longitud, donde se representa el
porcentaje de coincidencias interexónicas y intraexónicas de cada patron.
Podemos observar que a medida que el tamaño del patrón augmenta, este tiene
menos tendencia a localizarse intraexónicamente (y viceversa), pero
encontramos patrones que a pesar de su tamaño caen totalmente dentro de un
exón.
Se han considerado de interés aquellos patrones largos que se ubican
intraexónicamente en un porcentaje superior al 80% .
Se han despreciado los patrones que no cumplen esta condición porque
asumiendo que sólo se estan analizando los patrones largos, por azar tienen
tendéncia a localizarse interexónicamente.
De los datos seleccionados hasta el momento, sólo se consideran
significativos si se han encontrado un número considerable de veces dentro
de las secuencias. Para obtener este dato se tiene que recurrir a la tabla
inicial de resultados (tabla en Excel).
Se han despreciado los patrones que no cumplen esta condición porque, en
este caso, no es significativo un porcentaje sin tener en cuenta el número
de correspondencias del patrón dentro de las secuencias.
Una vez acabado este análisis, se han obtenido los siguientes patrones:
|
Tabla de resultados 2
| Longitud |
Patron |
Matches |
Descripcion |
| 17 |
PS00237 |
66 |
G-protein coupled receptors family. |
| 17 |
PS00034 |
14 |
�Paired box� domain. |
| 17 |
PS00179 |
20 |
Aminoacyl-transfer RNA synthetases classII. |
| 17 |
PS00408 |
11 |
Connexin 2. |
| 18 |
PS00338 |
14 |
Somatotropin, prolactin and related hormones. |
| 19 |
PS00280 |
12 |
Pancreatic trypsin inhibitor. |
| 21 |
PS00028 |
1777 |
Zinc finger C2H2 type domain. |
| 22 |
PS00029 |
895 |
Leucine zipper pattern. |
| 23 |
PS00260 |
12 |
Glucagon/GIP/Secretin/VIP family. |
| 24 |
PS00027 |
100 |
'Homeobox' domain. |
| 27 |
PS00031 |
30 |
Nuclear hormones receptors DNA-binding domain. |
| >27 |
PS00207 |
4 |
Transferrine signature. |
| >27 |
PS00347 |
4 |
Poly (ADP-ribose) polymerase zinc finger domain. |
| >27 |
PS00420 |
4 |
SRCR domain (proteína de células secretoras). |
| >27 |
PS00023 |
12 |
Type II fibronectin collagen-binding domain. |
|
Conclusiones...
En esta tabla estan representados todos los patrones de Prosite que cumplen
las siguientes características:
-Se han encontrado una o más veces dentro de alguna de las secuencias
analizadas
-Tienen un tamaño mayor que 17 aminoácidos (equivalente a 51 nucleótidos)
-Tienen una ubicación mayoritáriamente intraexónica, despreciándose
aquellos patrones que se han encontrado pocas veces en las secuencias.
|
VOLVER A LA TABLA DE CONTENIDOS
|
5.CONCLUSIÓN
Podemos observar que los 10 primeros patrones más abundantes ( tabla de resultados 1)
constituyen casi el 97% de los patrones encontrados. Estos patrones podemos
agruparlos en conjuntos más generales:
Patrones de fosforilación (61.43%): Constituyen el grupo más importante, dando a entender
que este sistema es, con diferencia, el proceso más habitual en
la célula, estos patrones se encuentran en proteínas implicadas
en rutas de transducción de señales, activación-inactivación,
señalización, etc.
Patrones de modificaciones post traduccionales
(33.37%): Este grupo incluye uno de los dominios de glicosilación,
proceso también muy importante ya que la gran mayoría de
proteínas humanas se N-glicosilan.
Patrones de unión (2.23%): En
este grupo encontramos otros dominios indispensables para la vida como
patrones de unión al DNA, patrones de unión a glicosaminoglicanos
(implicados en la formación de la estructuración pluricelular)
y patrones de unión a elementos ajenos al organismo.
De todos los patrones considerados significativos ( tabla de resultados 2) se
ha podido observar que 14 (93%) de estos patrones son
exclusivos de organismos eucariotas, y que 10 (67%) son característicos
de funciones típicamente pluricelulares como pueden ser comunicación
intercelular, digestión, regulación, desarrollo ontogénico,
estructuración...etc.
¿Se puede explicar por qué justamente estos patrones se
encuentran con una frecuencia mayor a la esperada en una ubicación
intraexónica? La siguiente hipótesis trata de explicarlo aunque se debería seguir investigando
para aportar mayor firmeza a los resultados:
Los genes más primitivos y básicos se formaron antes de la inserción de intrones y/o la unión de diferentes
exones para formar nuevas proteínas. Cada uno de estos genes monoexónicos tenía uno o
más dominios funcionales.
Cuando se produjo la unión de diferentes exones, los dominios funcionales
seguirían estando en una localización intraexónica. Es más tarde, cuando estos diferentes exones
adquieren una continuidad proteica, cuando se produce la aparición de nuevos dominios, que no
sólo se encontrarán en una localización intraexónica
(la presión selectiva recae finalmente sobre los aminoácidos de las proteínas
y por tanto, es independiente del lugar donde se encuentre el codón siempre
y cuando este acabe expresándose). Por tanto, es más probable
encontrar un gen relativamente nuevo (común a eucariotas o mamíferos) en una posición interexónica
que un gen antiguo (común a procariotas).
Es razonable también que muchos genes se salgan de esta regla simplemente por la complejidad misma del proceso (azar).
|
VOLVER A LA TABLA DE CONTENIDOS
Direcciones de contacto:

eulalia.belloc01@campus.upf.es
alba.duch01@campus.upf.es
sergio.lois01@campus.upf.es
alberto.moldon01@campus.upf.es
|
| |
