Options.
Scanner gives the user different using options:
- -h: to receive information about the usage of the program. Type 1 after "-h" for information in Spanish, 2 for Catalan, 3 for English. This option must be followed by a number.
- -v: to receive information about the program status while it's running.
- -m: to receive information about the matrix/es name, dimensions and consensus sequence.
- -s: to receive information about the sequence/s name, length, G+C.
Requirements.
The input files must follow some basic structural requierements.
Examples of sequences on FASTA format:
Back to Homepage
>name of seq 1
GTACGTACGTATCGTACGATCGATGC
TATAAGCATGCTAGCTGCAGCAGCAC
CTACGAT
>name of seq 2
CACTGCTAGCTTACGACGTCGATTAT
ACAGTGGCATCACTACGCGTAC
>name of seq 3
TAGTACTCGTAACGATAGCTACGACT
AGAGCGCG
Programming details.This program allows you to identify binding sites of transcriptional factors using weight matrixes.
- BASIC INSTRUCTIONS:
There are some options that the user should read before using it.
You must download it!
You must install it in a OS compatible with PERL.But, how does it work? There are several steps:
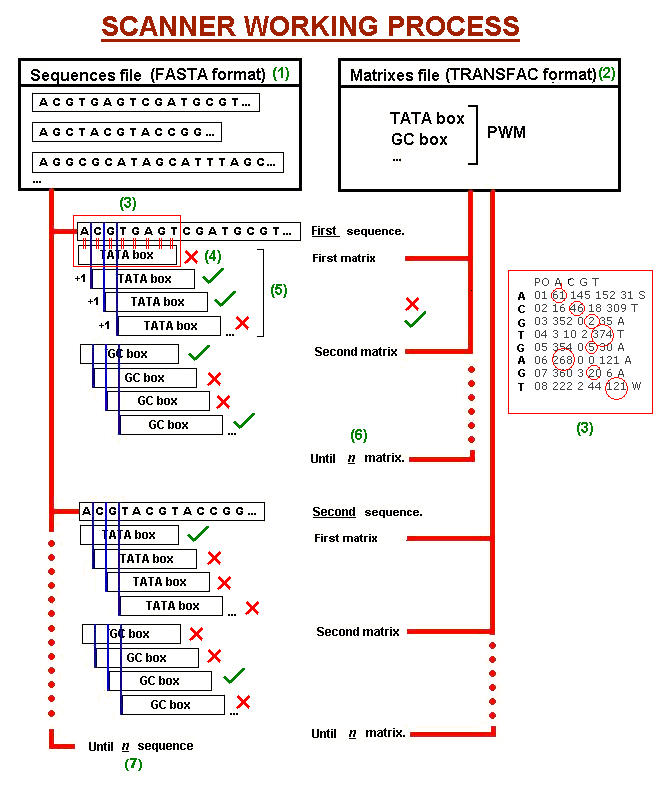
- Open the sequence/s file.(1)
- This file is splited sequence by sequence into an array.
- Every element of this array (a sequence) is processed:
- Delete the name (is saved in another array, which accumulates all the names).- Every rearranged sequence is an element of a new array that we will be using to score.
- Split the sequence line by line, join all the elements of this array again (so we delete the line change character).
- Open the matrix/es file.(2)
- This file is splited matrix by matrix into an array.
- Every element of this array (a matrix) is processed:
- Delete the name (is saved in another array, which accumulates all the names), the positions and the consensus (saved also in a new array).
- Split row by row, and each row is splited again as a new array. - Every element is operated to find the log-likelihood values. We use the expression:
Log-likelihood value= ln [(a/sum)/p] a = old matrix value.
sum = addition of the whole row values.
p = 'a priori' probability of any nucleotide (we take 0.25).- All these new values are saved in a three-dimensioned array (the third dimension is to determine the matrix they come from).
- Sequence candidates scoring.(3)
- We use the last sequences array arranged in the first step.
- We split the first element of this array (the first sequence) nucleotide by nucleotide.
- Comparison to the matrix:- The program draws a sliding window starting on the first position of the sequence. The width of this sliding window is the matrix number of rows.- This comparison is repeated for all the matrices (6) on each sequence (7).
- Every position of the sequence incluided in the current window is compared with the same matrix position and a log-likelihood score is assigned to the position according to the nucleotide.
- The total window score is the addition of each single position score.
- Sequence candidates filtering:(4)if the current window score is higher than the threshold, the content of the window is considered a binding site candidate.- This process is repeated moving the sliding window one position at a time (5), until the program cannot draw any more window on the sequence.
Fig.2: Scheme showing the SCANNER working process. For more details follow numbers on the text.
Back to Homepage
By: Guiomar Solanas & Alex Vendrell. Universitat Pompeu Fabra, March 2002.
For further information... Send an e-mail to the authors.