Identificació computacional de llocs d'unió
a factors de transcripció
Les següents instruccions us han de servir per desenvolupar un programa
en Perl al qual li doneu un fitxer amb matrius de factors de transcripció
(FTs) i un altre fitxer amb una seqüència d'una regió
promotora en format FASTA i el programa mostra la llista dels FTs proporcionats
amb un p-value per cada FT que ens indiqui la probabilitat de rebutjar
erròniament la hipòtesi inicial de que el factor no s'uneix.
Com a conjunt de matrius de FTs fareu anar el que trobareu dins el fitxer
humanfactors_selectedmatrices.txt
que conté matrius de 13 FTs humans diferents extretes del conjunt de
matrius de FTs que fa anar el programa
PROMO.
El programa, per tant, l'hauriem de poder cridar des del shell d'aquesta forma:
$ ./identificacio_de_lufs.pl humanfactors_selectedmatrices.txt sequenciapromotora.fa
Desenvolupeu el programa seguint els següents passos:
- comenceu fent que el programa llegeixi el fitxer de matrius, matriu a
matriu, enregistrant-la en memoria com a un hash de vectors. Per comprovar
que el programa llegeix i emmagatzema be les matrius, feu que us mostri
per pantalla el contingut del hash de vectors un cop l'heu omplert.
Cada matriu en el fitxer de matrius segueix el següent format:
FA AP-1 [T00029]
XX
P0 A C G T
00 3 0 7 28
01 4 1 31 2
02 38 0 0 0
03 0 0 38 0
04 0 0 0 38
05 0 38 0 0
06 38 0 0 0
//
on la primera columna de texte determina el tipus d'informació
emmagatzemada a la línia de la següent forma:
- FA nom del FT i número d'accés a la base de dades
de FTs TRANSFAC.
- XX línia separadora que marca el començament de la
matriu en sí mateixa.
- P0 encapçalament de la matriu indicant a quin nucleòtid
està associat cada columna de les línies següents.
- 00 occurrències de cada nuclòtid a la primera posició
del motiu.
- 01 occurrències de cada nuclòtid a la segona posició
del motiu.
- nn occurrències de cada nuclòtid a la nn-èssima
posició del motiu.
- // darrera línia de la informació relativa a la matriu.
- per a cada FT heu de transformar la seva matriu d'ocurrències de nucleòtids
en una matriu de pesos (popularment coneguda en anglès com a position weight
matrix) de forma que obteniu una matriu on cada nucleòtid té associat
un pes a cada posició diferent del motiu. Si denotem aquest pes per
wi(x) per indicar el pes associat al nucleòtid x a la
posició i-èssima dins un motiu que anomenarem M, el seu
càlcul el farem de la següent forma:
wi(x) = log(piM(x)) -
log(pB(x)),
on piM(x) és la proporció d'ocurrències
del nucleòtid x a la posició i del motiu M, i
pB(x) és la proporció d'ocurrències del
nucleòtid x al llarg de tota la seqüència promotora, es a dir,
la proporció de vegades que esperem observar el nucleòtid x
a l'atzar dins aquesta seqüència. En el cas de que
piM(x) sigui 0 eviteu fer el càlcul del seu
logaritme i considereu en el seu lloc un valor negatiu molt alt, com ara -999.
- per a cada FT heu de calcular la puntuació que proporciona la seva
matriu de pesos a cada possible posició d'unió del FT
dins la seqüència promotora. Es a dir, per una seqüència
promotora de llargada N i un FT que s'uneix a f nucleòtids,
on per tant la matriu de pesos te f posicions, hi ha N-f+1 llocs d'unió
possibles cadascun dels quals el puntuarem desplaçant una finestra de longitut
f al llarg de la seqüència:
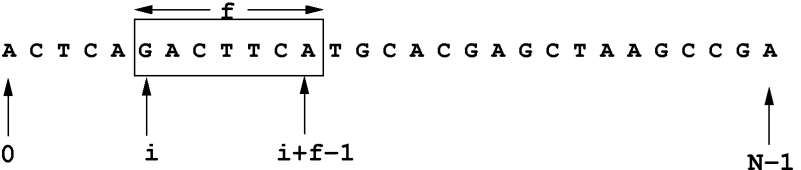
si denotem per siM(x) la puntuació (en anglès
score) que obtenim per al lloc d'unió que comença a la posició
i dins una seqüència de nucleòtids x utilitzant una
matriu de pesos associada al un motiu M, farem el seu càlcul de la
següent forma:
siM(x)=Σj=ii+f-1 wj(xj),
on xj denota el nucleòtid j-èssim dins la
seqüència x. Enregistreu la puntuació màxima que
obteniu al llarg de la seqüència així com la posició del
lloc d'unió que la proporciona. Denotem en aquest document la
puntuació màxima observada en la seqüència promotora
amb s*.
- finalment, per cada FT, després del càlcul anterior cal
estimar la freqüència amb la qual s'observa a l'atzar la
puntuació màxima antrior. Aquesta estimació és
una aproximació a la probabilitat de rebutjar erròniament
la hipòtesi inicial de que el factor no s'uneix (p-value).
Valors petits són indicatius de que la unió que identifiquem
es difícil d'observar a l'atzar i, per tant, es raonable pensar que
aquest FT s'uneix efectivament a la regió promotora. Aquest
càlcul el farem de la següent forma:
- generem una permutació dels nucleòtids de la regió
promotora mitjançant el següent algorisme (conegut com a
algorisme de Fisher-Yates):
assumim tenim la sequencia promotora en un vector @v, el seguent
algorisme permutara a l'atzar els elements de @v
my $n = scalar(@v);
my $i = $n - 1;
while ($i >= 0) {
my $j = int(rand($i+1));
if ($i != $j) {
my $tmp = $v[$i];
$v[$i] = $v[$j];
$v[$j] = $tmp;
}
$i = $i - 1;
}
- puntuem tots els llocs d'unió possibles al llarg de la
seqüència permutada enregistrant el valor màxim
observat
- repetim aquests dos passos 100 cops, comptant en quants d'aquests
casos la puntuació màxima observada és més
gran o igual que s*. Denotem aquest nombre de cops per
c, direm que el p-value associat al FT és igual
o més petit que c/100.
Cal tenir present que l'algorisme anterior és una aproximació
simplificada al problema i, per tant, d'una capacitat predictiva limitada.
Si voleu tenir una visió més real del problema d'identificar
computacionalment llocs d'unió a FTs és aconsellable que llegiu
els dos següents articles sobre el tema:
Wasserman WW, Sandelin A. Applied Bioinformatics for
the identification of regulatory elements. Nat. Rev. Genetics, 5(4):276-87, 2004.
Tompa et al., Assessing computational tools for the discovery of
transcription factor binding sites. Nat. Biotechnol., 23(1):137-44, 2005.