Burset and Guigó (Genomics 34, 353-367, 1996) attempted to develop data set and performance metrics standards for consistently testing of Gene Identification software. They tested a number of programs on a large set of vertebrate sequences with known gene structure, and several measures of predictive accuracy were computed at the nucleotide, exon, and protein product levels. The data set used by Burset and Guigó was highly biased including only relatively short DNA sequences with simple gene structure and high coding density. Thus, the performance of the programs is likely to provide an overoptimistic estimation of the actual accuracy of the progrmas when confronted with large DNA sequences from randomly sequenced genomic regions of less standard composition and more complex structure.
Recently, Rogic et al. (Genome Research 11, 817-832, 2001) published a new independent comparative analysis of seven gene prediction programs. The programs were again tested in single gene sequences--from human and rodent species. In order to avoid overlap with the training sets of the programs, only sequences were selected that had been entered in GenBank, after the programs were developed and trained.
The paper by Rogic et al. represented a valuable update on the accuracy of gene finders, but it suffered from the same limitation as did the previous work by Burset and Guig¾ and others: gene finders were tested in controlled data sets made of short genomic sequences encoding a single gene with a simple gene structure. These data sets are not representative of the genome sequences being currently produced: large sequences of low coding density, encoding several genes and/or incomplete genes, with complex gene structure. Thus, accuracy measures inferred from them are likely to be substantial overstimations of the actual accuracy of the programs in real genomic sequences. Indeed, the initial analysis on chromosome 22 suggests that the ab initio gene finders predict a large number of false positive genes, and while they tend to predict most of the known genes, only an small fraction of the genes have all exons correctly predicted. For instance, GENSCAN predicts 94% of the known genes in chromosome 22, but only about 20% of them had all the exons correctly predicted.
Ideally, one would like to benchmark the gene identification programs in real genomic sequences. The main problem is that until very recently on very few of these sequences the structure of the genes has been exhaustively verified by experimental means, making it almost impossible to construct large enough test sets of well characterized and large enough genomic sequences with which the accuracy of the gene predictions can be calibrated. Because of the lack of well characterized human sequences, and because genes or exons may remain undetected even if a detailed experimental analysis is performed, Guig¾ et al. (Genome Research 10, 1631-1642, 200) investigated the accuracy of ab initio gene finders by constructing a set of semi-artificial sequences to emulate large well annotated genomic sequences. In these semi-artificial sequences, known genic sequences have been embedded in simulated intergenic DNA. Therefore, the location of all coding exons is known.
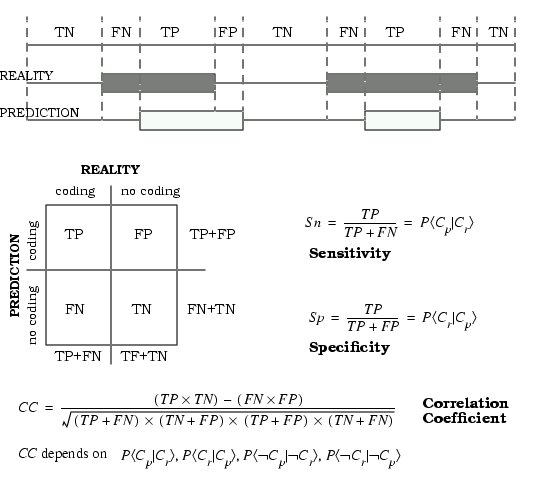
Prediction accuracy per base coding/non-coding.
Sn: Sensitivity = TP/(TP+FN)
TP:true positives
TN:true negatives
FP:false positives
FN:false negatives
Prediction accuracy with respect to exact prediction of exon start and end points
Sn: Sensitivity = fraction of correct exons over actual exons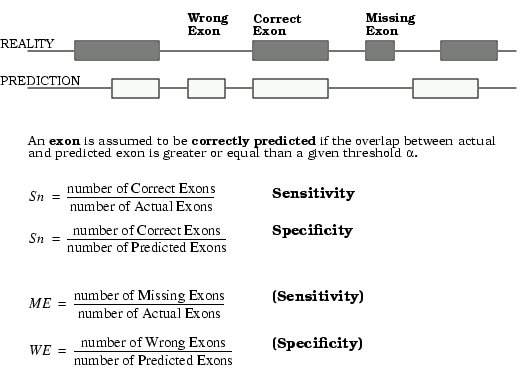
Predcition accuracy with respect to the protein product encoded by the predicted gene.
% Sim: percentage similarity between the amino acid sequence encoded by the predicted gene and the amino acid sequence encoded by the actual geneThe evaluation by Burset and Guigó consisted of three steps:
The ALLSEQ data set is characterized by two
features.
First, it includes only short sequences encoding a single
complete gene with simple structure, with high coding density,
and free of sequencing errors.
Second, ALLSEQ was built attempting to minimize the
overlap with the training sets used during the development of the
programs analyzed.
Thus, only sequences entered into the public databases
after January 1993 were selected into ALLSEQ. In addition,
a subset of ALLSEQ was considered
separately (NEWSEQ), comprising only those sequences that did not show a
significant similarity to the vertebrate sequences entered into the database
prior to January 1993. The ALLSEQ data set has 570 vertebrate sequences,
196 of them included in the NEWSEQ subset.
Files containing the DNA sequences in ALLSEQ, the locations of the exons in the sequences, and the corresponding encoded amino acid sequences can be found at http://genome.imim.es/databases/genomics96/index.html

| Nucleotide | Exon | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Program | dataset | Sn | Sp | CC | Sn | Sp | (Sn+Sp)/2 | ME | WE |
| fgeneh | allseq | 0.77 | 0.88 | 0.80 | 0.61 | 0.64 | 0.64 | 0.15 | 0.12 |
| newseq | 0.70 | 0.83 | 0.73 | 0.51 | 0.54 | 0.54 | 0.22 | 0.18 | |
| geneid | allseq | 0.63 | 0.81 | 0.65 | 0.44 | 0.46 | 0.45 | 0.28 | 0.24 |
| newseq | 0.58 | 0.78 | 0.60 | 0.41 | 0.43 | 0.42 | 0.34 | 0.27 | |
| geneparser2 | allseq | 0.66 | 0.79 | 0.65 | 0.35 | 0.40 | 0.37 | 0.29 | 0.17 |
| newseq | 0.63 | 0.76 | 0.62 | 0.33 | 0.39 | 0.36 | 0.32 | 0.20 | |
| genlang | allseq | 0.72 | 0.79 | 0.71 | 0.51 | 0.52 | 0.52 | 0.21 | 0.22 |
| newseq | 0.63 | 0.73 | 0.63 | 0.39 | 0.44 | 0.43 | 0.29 | 0.25 | |
| grail 2 | allseq | 0.72 | 0.87 | 0.76 | 0.36 | 0.43 | 0.40 | 0.25 | 0.11 |
| newseq | 0.69 | 0.85 | 0.72 | 0.34 | 0.41 | 0.38 | 0.30 | 0.13 | |
| sorfind | allseq | 0.71 | 0.85 | 0.72 | 0.42 | 0.47 | 0.45 | 0.24 | 0.14 |
| newseq | 0.65 | 0.79 | 0.65 | 0.36 | 0.39 | 0.38 | 0.29 | 0.19 | |
| xpound | allseq | 0.61 | 0.87 | 0.69 | 0.15 | 0.18 | 0.17 | 0.33 | 0.13 |
| newseq | 0.58 | 0.83 | 0.64 | 0.12 | 0.15 | 0.14 | 0.36 | 0.16 | |
| geneid+ | allseq | 0.91 | 0.91 | 0.88 | 0.73 | 0.70 | 0.71 | 0.07 | 0.13 |
| newseq | 0.88 | 0.87 | 0.84 | 0.68 | 0.64 | 0.66 | 0.10 | 0.15 | |
| geneparser3 | allseq | 0.86 | 0.91 | 0.85 | 0.56 | 0.58 | 0.57 | 0.14 | 0.09 |
| newseq | 0.83 | 0.89 | 0.82 | 0.50 | 0.53 | 0.51 | 0.17 | 0.09 | |
GeneID+ and GeneParser3 incorporate results from amino acid database searches to make the gene predictions.
Average CC for the programs analyzed in the ALLSEQ data set ranged from 0.65 to 0.78 at the nucletoide level, while the average exon prediction accuracy ranged from 0.37 to 0.64. Accuracy was lower and less variable between programs in the NEWSEQ dataset: average $CC$ ranged here from 0.60 to 0.70, and average exon accuracy ranged from 0.36 to 0.54. The authors of the study also found that the programs showed some dependency---usually only slight---on the phylogenetic group within vertebrates in which they had been trained and tended to perform worse on low G+C content sequences, but this was not a general trend. Programs were also sensitive to high rates of sequence errors, but their robustness varied widely.| Nucleotide | Exon | |||||||
|---|---|---|---|---|---|---|---|---|
| Program | Sn | Sp | CC | Sn | Sp | (Sn+Sp)/2 | ME | WE |
| fgenes | 0.86 | 0.88 | 0.83 | 0.67 | 0.67 | 0.67 | 0.12 | 0.09 |
| genemark.hmm | 0.87 | 0.89 | 0.83 | 0.53 | 0.54 | 0.54 | 0.13 | 0.11 |
| genie | 0.91 | 0.90 | 0.88 | 0.71 | 0.70 | 0.71 | 0.19 | 0.11 |
| genscan | 0.95 | 0.90 | 0.91 | 0.70 | 0.70 | 0.70 | 0.08 | 0.09 |
| hmmgene | 0.93 | 0.93 | 0.91 | 0.76 | 0.77 | 0.76 | 0.12 | 0.07 |
| morgan | 0.75 | 0.74 | 0.69 | 0.46 | 0.41 | 0.43 | 0.20 | 0.28 |
| mzef | 0.70 | 0.73 | 0.66 | 0.58 | 0.59 | 0.59 | 0.32 | 0.23 |
Other findings of Rogic et al. (2001) include dependency of some programs on G+C content, but little on phylogenetic specific training within mammals, and the general trend among the programs tested to have a very low proportion of correctly predicted short exons or very long exons. This, together with the fact that splice sites seem to be better predicted than start and stop codons, could explain the tendency, consistent among the programs, of better predicting internal exons than either internal or terminal exons. This, in turn, leads to rather poor predictions of the gene termini, as we will see later.
To overcame the lack of large well annotated genomic sequences in the human genome Guig¾ et al. constructed a set of semi-artificial genomic sequences. They proceeded in the following way. First, a typical benchmark set was extracted made of sequences from the EMBL database release 50 (1997). These are 178 human genomic sequences coding for single complete genes for which both the mRNA and the coding exons are known. We will refer to this set here as h178. The coding density of this set is 21%. Second, to simulate large sequence contigs, the sequences in h178 were randomly placed in a background of intergenic DNA, randomly generated using a Markov Model of order 5. The length of each artificial contig is generated randomly according to a normal distribution. Genomic fragments containing genes and random-sized segments of intergenic sequence are then concatenated until their combined lengths exceed the predetermined contig target length. The strands are also chosen at random for each genic subsequence. After this process, the 178 genic sequences collapsed into 42 semiartificial genomic sequences (SAG). We will refer to this set as Gen178. The coding densitiy on this set was only 2.3\%, very close now to the coding density estimated for the whole human genome.
The table below shows the accuracies of GENSCAN ---as the most accurate representative of "ab initio" gene finders---in the set of single gene sequences h178 and in the set of large artificial contigs Gen178.
| Nucleotide | Exon | Gene | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Program | dataset | Sn | Sp | CC | Sn | Sp | Sn+Sp/2 | ME | WE | MG | WG |
| genscan | Gen178 | 0.89 | 0.64 | 0.76 | 0.64 | 0.44 | 0.54 | 0.14 | 0.41 | 0.03 | 0.28 |
| h178 | 0.92 | 0.92 | 0.91 | 0.76 | 0.76 | 0.76 | 0.09 | 0.09 | |||
| blastx nr | h178 | 0.90 | 0.92 | 0.90 | 0.10 | 0.12 | 0.11 | 0.12 | 0.07 | ||
| genewise | Gen178 | 0.98 | 0.98 | 0.97 | 0.88 | 0.91 | 0.89 | 0.06 | 0.02 | ||
| h178 | 0.98 | 0.98 | 0.97 | 0.88 | 0.91 | 0.89 | 0.06 | 0.02 | |||
| procrustes | Gen178 | 0.93 | 0.94 | 0.93 | 0.80 | 0.75 | 0.77 | 0.10 | 0.16 | ||
| h178 | 0.93 | 0.95 | 0.93 | 0.76 | 0.82 | 0.79 | 0.11 | 0.04 | |||
| Very similar target | Moderate similar target | |||||||||||
| p-value < e-50 | e-50 < p-value < e-6 | |||||||||||
| 17 SAG sequences | 26 SAG sequences | |||||||||||
| Nucleotide | Exon | Nucleotide | Exon | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Program | Sn | Sp | CC | Sn | Sp | (Sn+Sp)/2 | Sn | Sp | CC | Sn | Sp | (Sn+Sp)/2> |
| genscan | 0.91 | 0.66 | 0.77 | 0.67 | 0.46 | 0.56 | 0.91 | 0.61 | 0.74 | 0.67 | 0.43 | 0.55 |
| genewise | 0.99 | 0.99 | 0.99 | 0.90 | 0.93 | 0.91 | 0.68 | 0.98 | 0.81 | 0.46 | 0.63 | 0.54 |
| procrustes | 0.92 | 0.96 | 0.94 | 0.80 | 0.75 | 0.77 | 0.66 | 0.79 | 0.72 | 0.48 | 0.32 | 0.40 |
Roderic Guig¾ (i Serra), IMIM and UB. rguigo@imim.es