Bioinformàtica. UPF
Let's perform a very simple exercise: given a nucleotide sequence, compute the number of times that the nucleotide A (Adenine) appears at a distance k from another nucleotide A. And let's do that for every possible k, from 0 to the length of the sequence. For instance if the sequence is
TAAGAGACTCATAAGT
these numbers are
K 0 2 1 3 2 2 3 2 4 1 5 2 6 1 7 2 8 2 9 1 10 2 11 1 12 0 13 0 14 0 15 0
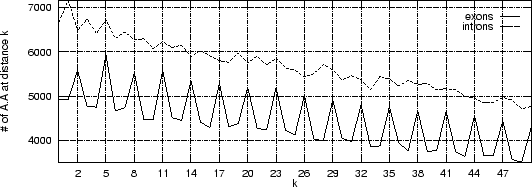
Let's repeat this exercise now for about 500 exon and 500 intron human sequences (actually only 200 bp taken from each exon, and each intron), and let's plot the cumulative frequency of occurrency of pairs A ... A at each possible distance k.
As it is possible to see, a clear periodic pattern arises from the set of exon sequences. The nucleotide A is more likely to be found at distance k=2,5,8, ... from another A than at other distances. This periodic pattern is absent in the intronic sequences.
Note that nucleotide pairs at a distance of k=2,5,8, ... nucleotides, are at the same codon position, whereas nucleotide pairs at other distances, are not.
This periodic pattern reflects the fact that proteins use the different amino acids with different frequencies, and that synonymous codons are used with different frequencies to code for a given amino acid. This causes coding sequences to exhibit an strong codon bias, which is (mostly) absent in non-coding sequnces. The codon bias causes the periodic pattern observed in coding sequences. This periodic pattern is characteristic of the 16 pairs of nucleotides, and not only of the pair A ... A.
Thus, measuring the strength of the periodic pattern in a sequence problem, we can measure the likelihood of the sequence being coding. A measure of DNA sequence periodicity is what we will call here a sequence coding statitic.
A coding statistic or codig measure can be defined as a function that computes given a DNA sequence a real number related to the likelihood that the sequence is coding for a protein.
Since the early eighties, a great number of coding statistics have been published in the literature. Most such coding statistics measure either codon usage bias, base compositional bias between codon positions, or periodicity in base occurrence (or a mixture of all them).
Below the human codon usage table.
The table can be used to estimate the likelihood of a sequence coding for a protein.
Indeed, by comparing the frequency of codons in a region of an species genome read in a given frame with the typical frequency of codons in the species genes, it is possible to estimate a likelihood of the region coding for a protein in such a frame.
Regions in which codons are used with frequencies similar to the typical species codon frequencies are likely to code for genes. This idea was first introduced by Staden and McLahlan staden:1982a. In the practice, the likelihood can be computed in a number of different ways. Here we compute it as a log-likelihood ratio.
Let ![]() be the
frequency (probability) of codon
be the
frequency (probability) of codon ![]() in the genes of the species under
consideration (from the codon usage table above)
in the genes of the species under
consideration (from the codon usage table above)
Then, given a sequence of codons
![]() , and assuming independence between adjacent codons
, and assuming independence between adjacent codons
For instance, if ![]() is the sequence
S=AGGACG,
when read in frame 1, it results in the sequence of codons
is the sequence
S=AGGACG,
when read in frame 1, it results in the sequence of codons
![]() ,
,
![]() .
.
Then
On the other hand, let ![]() be the frequency of
codon
be the frequency of
codon ![]() in a non-coding sequence.
in a non-coding sequence.
Assuming the random model of coding DNA,
![]() for all codons, and
for all codons, and ![]() for the above sequence of codons
for the above sequence of codons ![]() would be
would be
That is, the codons AGG and ACG are less common than expected in protein coding sequences. This makes rather unlikely (but not impossible) that this sequence codes for a protein in this particular frame.
In the practice, we compute a log-likelihood ratio.
The log-likelihood ratio for ![]() coding in frame
coding in frame ![]() ,
, ![]() ,
is
,
is
The log-likelihood ratios for ![]() coding in frames
coding in frames ![]() , and
, and ![]() (
(![]() and
and ![]() ) are computed in a similar way. Next above
log-likelihood ratios in the three frames computed on a real exon, and on a real intron sequence.
) are computed in a similar way. Next above
log-likelihood ratios in the three frames computed on a real exon, and on a real intron sequence.
| exon sequence | intron sequence | ||||
| coding frame | non coding frames | frame 1 | frame 2 | frame 3 | |
| 24.06 | -16.13 | -3.16 | -14.36 | -23.74 | -19.67 |
As it can be seen, in this case
the log-likelihood ratio ![]() is indeed greater than zero in the
coding frame of the exon sequence, while is smaller than zero in
the non-coding frames of the exon sequence and in all frames of the intron
sequence.
is indeed greater than zero in the
coding frame of the exon sequence, while is smaller than zero in
the non-coding frames of the exon sequence and in all frames of the intron
sequence.
The distribution of the scores of the Codon Usage log-likelihood ratios in the larger sets of intron and exon sequences are shown below
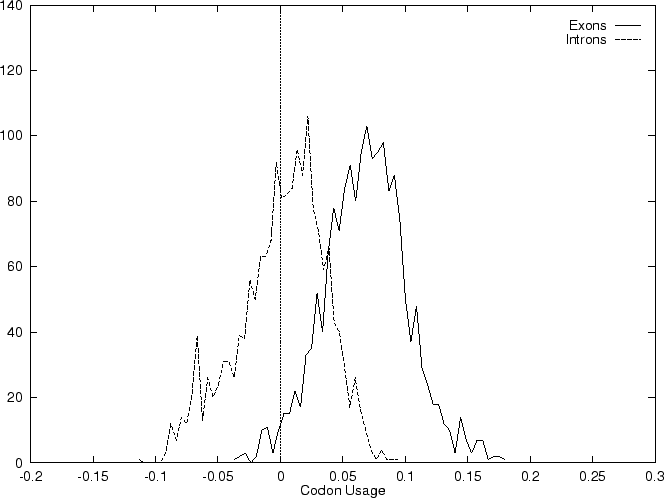
As it is possible to see, although the distributions are clearly distinct, there is substantial overlap between the Codon Usage scores in the sets of intron and exon sequences. As we will see, this is a general situation for all coding statistics.
In the practice, the problem is not usually to determine the likelihood that a given sequence is coding or not, but to locate the (usually small) coding regions within large genomic sequences. The typical procedure is to compute the value of a coding statistic in successive (usually overlapping) windows (an sliding window), and record the value of the statistic for each of the windows. This generates a profile along the sequence in which peaks may point to the coding regions and valleys to the non-coding ones.
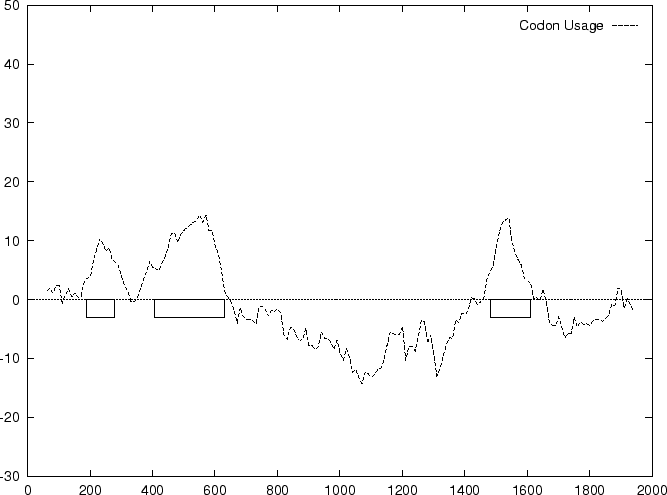
Below, we plot the result of sliding a window of length 120 bp, the
distance between consecutive windows being 10 bp, computing ![]() in
the three different frames, and plotting the highest value
obtained. The test sequence used is 2000 bp genomic region coding for
the human
in
the three different frames, and plotting the highest value
obtained. The test sequence used is 2000 bp genomic region coding for
the human ![]() -globin gene. In this case, the codon usage
log-likelihood profile reproduces fairly well the exonic structure of
this gene
-globin gene. In this case, the codon usage
log-likelihood profile reproduces fairly well the exonic structure of
this gene
Search by Content. Adapted from Guigo, R. ``DNA Composition, Codon Usage and Exon Prediction'' in Bishop M. ed , GENETIC DATABASES, Academic Press, 1999.
| rguigo@imim.es |