
MATERIALES Y MÉTODOS: MÓDULO I
MAPA CONCEPTUAL
|
|||
|

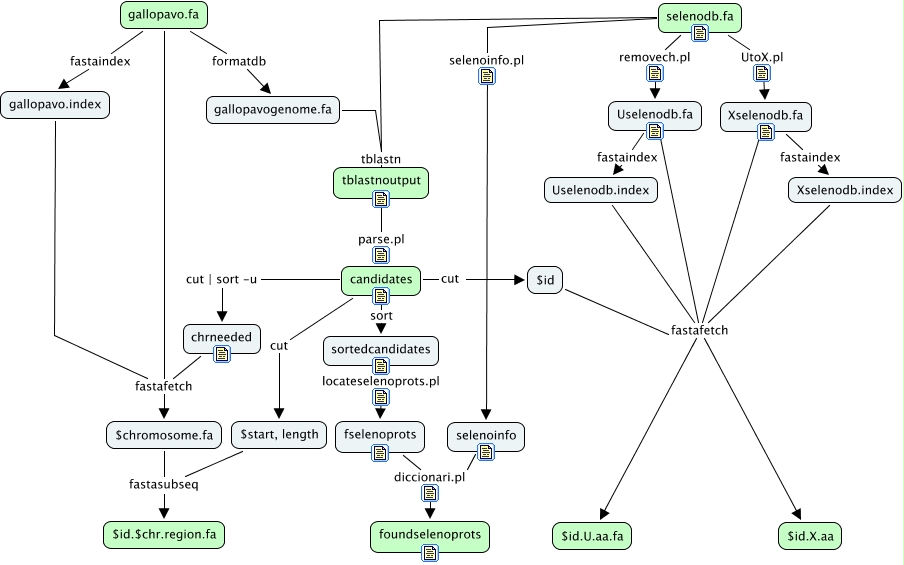
En este módulo partimos de la secuencia del genoma de Meleagris gallopavo i de un multifasta que contiene todas las selenoproteínas de SelenoDB. A las selenoproteínas les quitamos los caracteres no alfanuméricos y sustituímos las Us por Xs. En primer lugar, hacemos un tblastn de todas estas selenoproteínas contra el genoma de M. gallopavo. En un inicio tenemos un hit para cada exon pero parseamos el output del blast para quedarnos solamente con el inicio y el final de la región del genoma que ha dado homología con cada proteína (candidates). Hacemos fastasubseq para cada una de las regiones dando un margen de 15kb por ambos lados. Vemos que existen hits redundantes por que hay proteínas homólogas y parálogas que dan hit en la misma región del genoma. Para eliminar esta redundancia agrupamos todas las proteínas en función de la región del genoma en la que dan hit. De este modo pasamos de 331 hits a 36 regiones candidatas a albergar una selenoproteína (fselenoprots). Ademas añadimos una columna con la familia a la que pertenece cada selenoproteína (founselenoprots). Hacemos un fastafetch para cada proteína que nos ha dado hit.
BASH script
###############################################################################################
#################################### - MÓDULO 1 - ###################################
###############################################################################################
##################################### PRE-ANÁLISIS ####################################
###############################################################################################
#!/bin/bash
#$ -o our.stdout
#$ -e our.stderr
#$ -q llicen.q
#$ -N OurJob
#$ -cwd
export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH
cp /cursos/BI/bin/ncbiblast/.ncbirc ~/
export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH
export PATH=/cursos/BI/bin:$PATH
export WISECONFIGDIR=/cursos/BI/soft/wise-2.2.0/wisecfg
######CREACIÓN ÁRBOL DE DIRECTORIOS######
mkdir ~/gallopavodb
mkdir ~/gallopavodb/selenoprots
mkdir ~/gallopavodb/selenoprots/Uselenoprots
mkdir ~/gallopavodb/selenoprots/Xselenoprots
mkdir ~/gallopavodb/chromosomes
mkdir ~/gallopavodb/regions
mkdir ~/our_results
mkdir ~/our_results/exonerate
mkdir ~/our_results/exonerate/aa
mkdir ~/our_results/genewise
mkdir ~/our_results/genewise/aa
mkdir ~/our_results/proteins
mkdir ~/our_results/gallopavo_selenoproteins
mkdir ~/our_results/SECIS
mkdir ~/perlscripts/scripts_output
mkdir ~/perlscripts/scripts_output/t_coffee
mkdir ~/perlscripts/scripts_output/web
mkdir ~/perlscripts/scripts_output/web/Seleno_results/
cp ~/selenodb.fa ~/gallopavodb/selenoprots/selenodb.fa
cp ~/gallopavo.fa ~/gallopavodb/gallopavo.fa
###############################################################################################
########################FORMATEO GENOMA Meleagris_gallopavo y BASE DE DATOS SELENODB###########
formatdb -i ~/gallopavodb/gallopavo.fa -p F -n ~/gallopavodb/gallopavogenome.fa
fastaindex ~/gallopavodb/gallopavo.fa ~/gallopavodb/gallopavo.index
#removech.pl > Elimina los carácteres no alfanuméricos de las secuencias de SELENODB
~/perlscripts/removech.pl < ~/gallopavodb/selenoprots/selenodb.fa > ~/gallopavodb/selenoprots/Uselenoprots/Uselenodb.fa;
###Indexar el multifasta que contiene las proteínas de Selenodb (con U en Sec) ####
fastaindex ~/gallopavodb/selenoprots/Uselenoprots/Uselenodb.fa ~/gallopavodb/selenoprots/Uselenoprots/Uselenodb.index
#UtoX.pl > Cambia las U por X, para poder realizar la predicción con Exonerate
~/perlscripts/UtoX.pl < ~/gallopavodb/selenoprots/selenodb.fa > ~/gallopavodb/selenoprots/Xselenoprots/Xselenodb.fa
###Indexar el multifasta que contiene las proteínas de Selenodb (con X en Sec)###
fastaindex ~/gallopavodb/selenoprots/Xselenoprots/Xselenodb.fa ~/gallopavodb/selenoprots/Xselenoprots/Xselenodb.index
########################################### TBLASTN #########################################
blastall -p tblastn -i ~/gallopavodb/selenoprots/selenodb.fa -m8 -e1e-3 -d ~/gallopavodb/gallopavogenome.fa -o ~/gallopavodb/tblastnoutput;
################################## FILTRAJE INFORMACIÓN ##################################
# "chrneeded": fichero con los cromosomas en los que TBLASTN ha dado "hit".
cut -f2 ~/perlscripts/scripts_output/candidates | sort -u > ~/perlscripts/scripts_output/chrneeded
#selenoinfo.pl > Crea "selenoinfo": fichero con (ID prot) (Família Selenoprot.) (Especie). Se usará más adelante.
~/perlscripts/selenoinfo.pl < ~/gallopavodb/selenoprots/selenodb.fa | cut -c2-|sort|uniq > ~/perlscripts/scripts_output/selenoinfo;
#parse.pl > Parseado del output del TBLASTN. Cada proteína nos da distintos hits que corresponden
#a distintos exones. Crea un único hit con las posiciones más extremas y los valores que le pondremos al fastasubseq
#($start=$ini-20000), $lenght=$fin-$start+20000. También decide si el hit está en la cadena sense o antisense (-trev);
#Candidates fichero con (ID prot) (chr) (inicio) (fin) (start) (lenght) (-trev)
~/perlscripts/parse.pl < ~/gallopavodb/tblastnoutput | tail -n +2 | sort -g -k2 > ~/perlscripts/scripts_output/candidates;
sort -n -k2 -n -k3 ~/perlscripts/scripts_output/candidates > ~/perlscripts/scripts_output/sortedcandidates;
# "sortedcandidates" > Organización hits según región del genoma de 'Meleagris gallopavao' (k2 = chr; k3 = inicio)
#locateselenoprots.pl > Detectamos que hay hits en regiones muy próximas y solapantes en el genoma. Asumimos que todos
#los hits que caen en una misma región del genoma nos informan sobre una sola posible selenoproteína. Empíricamente
#determinamos que un rango de 75kb a cada lado nos permite agrupar todos los hits que nos informan sobre una sola proteína
#sin que se solape con ninguna otra. Reducimos la cantidad de candidatos de 231 a 36.
~/perlscripts/locateselenoprots.pl < ~/perlscripts/scripts_output/sortedcandidates > ~/perlscripts/scripts_output/fselenoprots;
#En "fselenoprots" cada proteína recibe un nuevo identificador (Seleno(nº)).
#Además de la información de 'sortedcandidates'.
#diccionari.pl > Añade la información obtenida con el programa selenoinfo.pl a partir de los nombres del archivo multifasta
#que contiene todas las selenoproteínas de SelenoDB. En el programa diccionari.pl llenamos un hash a partir de la información
#del documento selenoinfo. ATENCIÓN: diccionari.pl llena el hash desde DATA, que está al final del documeno, despues de __END__,
#y contiene el contenido de "selenoinfo". Esto se debe cambiar si se usa una query distinta.
~/perlscripts/diccionari.pl < ~/perlscripts/scripts_output/fselenoprots > ~/perlscripts/scripts_output/foundselenoprots;
# "foundselenoprots": Fichero con (SelenoX) (Chr) (inicio) (fin) (ID prot) (família selenoprot) (espécie)
#IMPORTANTE: vemos que todas las proteínas que hemos agrupado dentro de cada región pertenecen a la misma familia con la
#excepción de las proteínas que tienen varios parálogos. Esto nos da confianza y nos permite saber a qué família podría
#pertenecer cada proteína predicha.
######################################### FASTAFETCH #########################################
#Passamos de multifastas a archivos individuales con las proteínas y cromosomas que necesitamos para posteriores análisis.
### PROT U SELENODB ###
n=$(wc -l ~/perlscripts/scripts_output/candidates | cut -d ' ' -f1);
for line in $(seq 1 $n); do
id=$(head -$line ~/perlscripts/scripts_output/candidates | tail -1 | cut -f1 | tr -d ' ');
fastafetch ~/gallopavodb/selenoprots/Uselenoprots/Uselenodb.fa ~/gallopavodb/selenoprots/Uselenoprots/Uselenodb.index $id > ~/gallopavodb/selenoprots/Uselenoprots/$id.U.aa.fa;
done;
### PROT X SELENODB ####
n=$(wc -l ~/perlscripts/scripts_output/candidates| cut -d ' ' -f1);
for line in $(seq 1 $n); do
id=$(head -$line ~/perlscripts/scripts_output/candidates | tail -1 | cut -f1 | tr -d ' ');
fastafetch ~/gallopavodb/selenoprots/Xselenoprots/Xselenodb.fa ~/gallopavodb/selenoprots/Xselenoprots/Xselenodb.index $id > ~/gallopavodb/selenoprots/Xselenoprots/$id.X.aa.fa;
done;
### CROMOSOMAS ####
for chromosome in $(cat ~/perlscripts/scripts_output/chrneeded); do
echo "fasta fetch for chromosome: $chromosome ";
fastafetch ~/gallopavodb/gallopavo.fa ~/gallopavodb/gallopavo.index $chromosome
> ~/gallopavodb/chromosomes/$chromosome.fa;
done;
######################################### FASTASUBSEQ #########################################
#Con fastasubseq extraemos las regiones candidatas a contener una selenoproteína. En las columnas 5 y 6 de candidates
#se encuentran respectivamente los argumentos $start y $length. En parse.pl, cuando $start era menor de 1 le asignábamos
#el valor de 1 pero no sabíamos si la posición final estaría dentro o fuera del cromosoma o del scaffold. Si fastasubseq
#intenta extraer una secuencia que va más allá del final, te da un error que te informa de la última posición del cromosoma
#o el scaffold ('must end before...'). Redirigimos el standar error a un archivo y, si está lleno, extraemos la última
#posición, recalculamos la longitud y repetimos el fastasubseq.
n=$(wc -l ~/perlscripts/scripts_output/candidates | cut -d ' ' -f1);
for line in $(seq 1 $n); do
chr=$(head -$line ~/perlscripts/scripts_output/candidates | tail -1 | cut -f2 | tr -d ' ');
start=$(head -$line ~/perlscripts/scripts_output/candidates | tail -1 | cut -f5);
length=$(head -$line ~/perlscripts/scripts_output/candidates | tail -1 | cut -f6);
id=$(head -$line ~/perlscripts/scripts_output/candidates | tail -1 | cut -f1 | tr -d ' ');
fastasubseq ~/gallopavodb/chromosomes/$chr.fa $start $length > ~/gallopavodb/regions/$id.$chr.region.fa 2> ~/perlscripts/scripts_output/error_fastasubseq_regions;
error=$(cat ~/perlscripts/scripts_output/error_fastasubseq_regions);
if [ "$error" != "" ]; then
end_of_seq=$(cat ~/perlscripts/scripts_output/error_fastasubseq_regions | egrep 'must end before'| cut -d '(' -f2 | cut -d ')' -f1);
length=$(( $end_of_seq - $start ));
fastasubseq ~/gallopavodb/chromosomes/$chr.fa $start $length > ~/gallopavodb/regions/$id.$chr.region.fa;
fi;
done;

......................................................................................................................................................................................................................................................