Discusión
Esquema de los resultados obtenidos
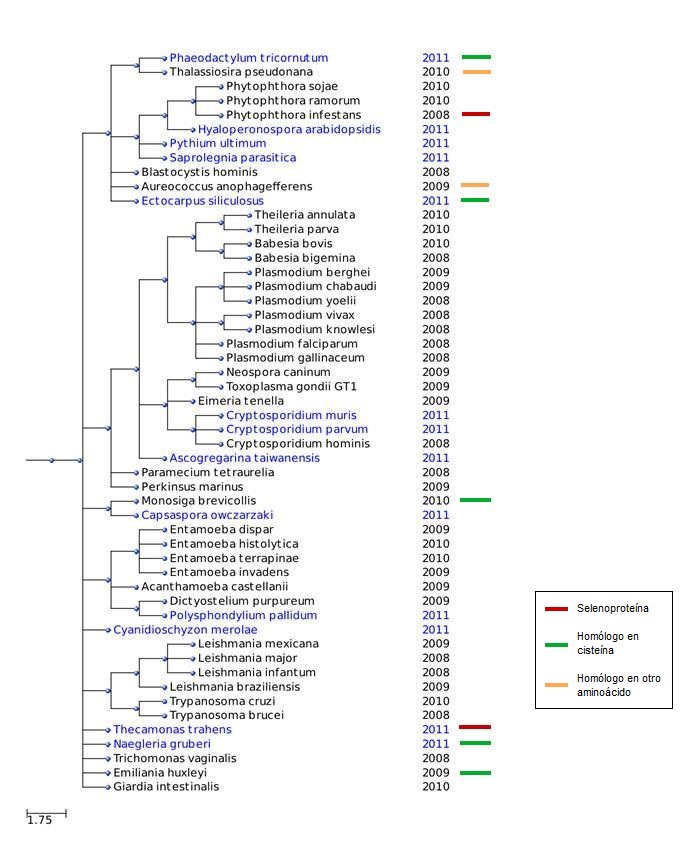{kind=link}
tBLASTn
Al hacer los tBLASTn de forma manual usamos la query de Ciona intestinalis para dsbA y la query humana para SelI. En dsbA obteníamos muy pocos hits con E-values significativos, es decir, menores a 0'001, por lo que sólo continuamos analizando aquellos hits con buenos E-values. Para SelI obtuvimos muchos hits con E-values significativos y en contigs diferentes.
Al analizar los tBLASTn en dsbA, algunas U alineaban con una C o con un gap de modo que indicaba que ahí teníamos un posible homólogo con cisteína o una posible selenoproteína. Estos hits los seguimos analizando.
En los tBLASTn de SelI, ninguno de los hits obtenidos presentaba alineada la región de la query que contiene la selenocisteína. A pesar de ello, seguimos analizando el mejor hit de cada contig, aquel que alineaba una región amplia y que además tenía un buen E-value (algunos hits con buenos E-values alineaban regiones muy pequeñas de la query, por eso los descartamos).
En cuanto a los tBLASTn hechos de forma automática, analizamos los genomas de los años 2011 usando como queries de dsbA Ciona intestinalis, Arthroderma gypseum y Psychromonas ingrahamii. Los resultados obtenidos para dsbA con la query de C.intestinalis los comparamos con los tBLASTn manuales y vimos que eran los mismos. Para SelI analizamos de forma automática los genomas de 2011 usando como queries selenoproteínas de Homo sapiens, Mus musculus y Phytophora infestans. En este caso también obtuvimos los mismos resultados de forma manual y automática.
Al ver que la automatización daba los mismos resultados que los obtenidos manualmente, decidimos analizar de forma automática los genomas de los años 2010, 2009 y 2008 y también la maquinaria para los genomas de 2011.
Fastasubseq
En el fastasubseq manual cogíamos una longitud de 5000 nucleótidos. Para determinar dónde empezar a coger la región que contiene nuestro gen, restamos unos 2000 nucleótidos al nucleótido de inicio. En el fastasubseq automático ampliamos la región a extraer para asegurarnos que en medio estaba la secuencia que nos interesaba. En este caso, al nucleótido de inicio le restamos 20000 nucleótidos y extraemos una región de 50000 nucleótidos.
Como pega habría que señalar que algunos contigs eran demasiado pequeños de modo que al hacer el fastasubseq, la región a extraer tenía que ser menor a 5000 nucleótidos, o no podíamos conseguir que nuestra secuencia de interés estuviese en el centro de la región extraída. En el método automático, para prevenir este problema, ideamos un sistema con el cual, en una secuencia contig pequeña, partíamos de un inicio de secuencia de 0 (start).
Exonerate y Genewise
En algunos de los hits analizados no obtuvimos un buen resultado usando el Exonerate de modo que tuvimos que recurrir a usar el Genewise. En estos genomas, los pasos anteriores al Exonerate eran correctos pero el Exonerate estaba vacío. Usando el Genewise obtuvimos resultados positivos en algunos casos.
Al hacerlo de forma manual, si no obteníamos resultado usando el Exonerate, usábamos seguidamente el Genewise. Sin embargo, al hacer el análisis de modo automático, usamos ambos métodos y sólo consultamos el Genewise en caso de que falle el Exonerate.
Fastatranslate
Para traducir los nucleótidos en aminoácidos usamos el Fastatranslate, que traduce la secuencia nucleotídica obtenida con el FastaseqfromGFF. El resultado del fastatranslate contiene los 6 posibles ORFs, de entre los cuales escogemos el más adecuado, aquel que contenga menos asteriscos (correspondientes a codones de stop). Para comprobar que el resultado es correcto o, en caso de obtener un empate en el número de asteriscos, para escoger el translate idóneo, hacemos un BLASTp recíproco contra la base de datos del NCBI para ver si obtenemos la selenoproteína de partida como hit y de este modo cerrar el círculo.
En el caso del paso automatizado del fastatranslate, hemos introducido la opcion -F 1 que selecciona la mejor proteína, aquella con menos asteriscos y los comparamos con los resultados obtenidos de forma manual.
Cuando todas las proteínas traducidas contenían más de 5 asteriscos, seguimos con el análisis pero no obtuvimos ningún resultado positivo.
Tcoffee
Al analizar los Tcoffees podemos ver si la selenocisteína de la query alinea con una cisteína o con un gap. En el caso de que la U de la query alinee con una C diremos que hemos obtenido un homólogo en cisteína, cuando alinea con un gap, vemos si la región circundante está conservada y si lo está decimos que en ese genoma tenemos una posible selenoproteína. Para comprobar que realmente contiene una selenoproteína miramos en el archivo obtenido con Exonerate si el gap que alinea con la U contiene el codón TGA.
En Arthroderma gypseum y Psychromonas ingrahamii, las queries no son selenoproteínas sino que son homólogos en cisteína ya que en lugar de tener una U tienen una C.
SECIS
Para ver si los protistas analizados contienen elementos SECIS, usamos el programa de la web de bioinformática en el que introducimos el fastasubseq de cada especie contra cada semilla.
También automatizamos la búsqueda de elementos SECIS usando el programa perl que nos facilitaban en la web. Inicialmente hicimos una búsqueda muy permisiva y encontramos SECIS en casi todos los genomas. Como esto no correspondía con los resultados obtenidos de forma manual, hicimos otro programa que buscaba SECIS usando los parámetros predefinidos.
Finalmente nos basamos en los resultados que obtuvimos usando la página web sólo para aquellos protistas en los que habíamos encontrado selenproteína.
No siempre hemos encontrado SECIS en aquellos genomas que probablemente contienen selenoproteínas. En algunos casos estos genomas carecen de elementos SECIS mientras que, sin embargo, hay SECIS en genomas en los que no hemos encontrado ni selenoproteínas ni homólogos con cisteína.
En un principio buscamos elementos SECIS en protistas que carecen de selenoproteínas, tanto en aquellos en los que hemos encontrado homólogos en cisteína como en los que no hemos encontrado nada, pero no mostramos estos resultados porque consideramos que si el protista no contiene selenoproteína no tiene sentido indicar si contiene elementos SECIS, ya que son un elemento necesario para que la maquinaria reconozca el codón UGA como una selenocisteína y no como un codón de STOP.
Muchos de los métodos de identificación de selenoproteínas se basan en la prediccioón de elementos SECIS en los genomas de los organismos. El problema yace en que es posible que la predicción de SECIS nos dé algún falso positivo o algún falso negativo. En éste último caso sería posible que en realidad ese organismo presentase este elemento en su genoma. Si nuestra búsqueda de selenoproteínas se basase en esta predicción estarímos cometiendo numerosos errores, por eso también hacemos los demás análisis.
La ausencia de elementos SECIS detectables en la región genómica downstream en aquellos organismos que presentan una selenoproteína puede ser debida a caracterícas inusuales de ese elemento SECIS, a la presencia de intrones largos o de numerosos intrones en la región 3'UTR o a la baja calidad del ensamblaje en el genoma. Por este motivo, seguiremos considerando que, a pesar de no encontrar elementos SECIS, algunos protistas tienen selenoproteínas en sus genomas.
BLAST recíproco
Para cerrar el círculo hemos hecho un BLASTp recíproco de la secuencia proteica obtenida contra la base de datos del NCBI. De este modo comprobamos si con nuestra proteína traducida encontramos homología con la selenoproteína analizada. En el caso de no encontrar la selenoproteína de partida, descartamos el resultado como una posible selenoproteína y lo consideramos como un resultado negativo.