Laura Selva Jové y Ana Sesé Nadal
Facultad de Ciencias de la Salud y de la Vida
Universidad Pompeu Fabra
En este trabajo se hace un estudio genómico computacional de la proteína de fusión FUS-CHOP. Esta proteína es el resultado de una translocación cromosómica t(12;16)(q13;p11), que involucra a los genes FUS (TLS) y CHOP (DDIT3), y está relacionada con el liposarcoma mixoide. A través de este estudio se caracterizará la estructura exónica, homología, función y expresión de los genes FUS y CHOP por separado. También, se hará un estudio sobre la región promotora y los factores de transcripción involucrados en la regulación de la expresión de los genes. Además de analizar los genes por separado, se hará un pequeño análisis sobre la proteína de fusión FUS-CHOP.
In this project is realized a computational genomic study of the FUS-CHOP fusion protein. This protein is the result of a characteristic chromosomal translocation, t(12;16)(q13;p11) that involve FUS(TLS) and CHOP(DDIT3) genes, and the translocation is related to myoxid liposarcoma. Throughout this study will be characterized the exon structure, the homology, the function and the expression of FUS and CHOP genes. Also, a study of the promotor region and transcription factors involved in the gen expression regulation will be done. Besides of analizing FUS and CHOP genes separately, it is analized the FUS-CHOP fusion protein and its function.
Sinónimos: DDIT3 (DNA damage-inducible transcript 3), GADD153 (Growth arrest and DNA-damage-inducible protein), C/EBP-homologous protein.
Estructura: La proteína CHOP está codificada por el gen DDIT3 (ENSG00000175197), que se encuentra en el cromosoma 12(q13.1-q13.2), concretamente en la región 56,196,640-56,200,567.

Figura 1. Localización cromosómica de DDIT3
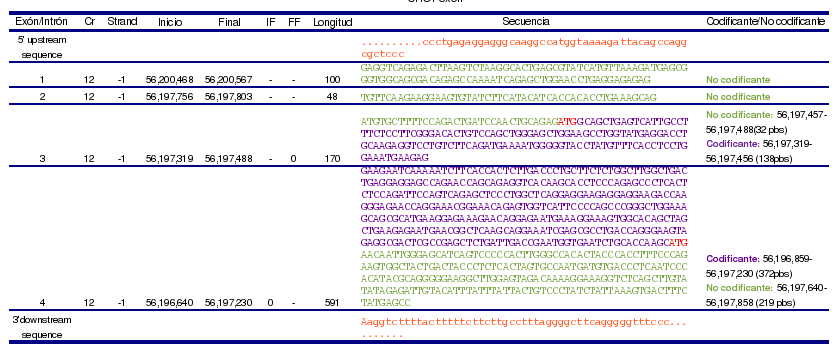
Este gen contiene cuatro exones, de los cuales, los exones 1 y 2 no son codificantes, y los exones 3 y 4, son parcialmente codificantes, ya que contienen la región 5' UTR y 3' UTR, respectivamente. Dan lugar a un solo transcrito (ENST00000346473) que tiene un tamaño de 909 pares de bases, y está formado por 169 aminoácidos.
![]()
Figura 2. Estructura de los exones de DDIT3
Función: La proteína CHOP actúa como inhibidor de la unión al DNA de los factores de transcripción C/EBP y LAP. CHOP inhibe la actividad de unión al DNA de estos factores mediante la formación de heterodímeros que no podrán unirse al DNA.
Familia: pertenece a la familia bZIP
Subunidad: Heterodímero
Localización subcelular: Nuclear
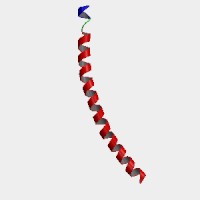
Figura 3. Estructura tridimensional de la proteína CHOP
Sinónimos: TLS (Translocated in liposarcoma)
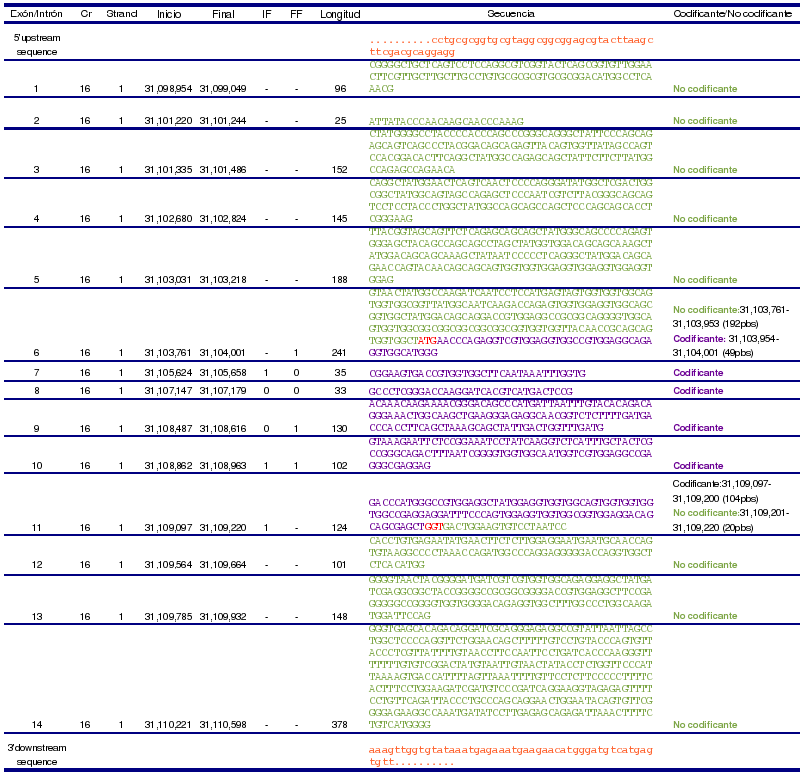
Estructura: La proteína FUS está codificada por el gen FUS (ENSG00000089280), que se encuentra en el cromosoma 16(p11.2), en la región 31,098,974-31,110,424.

Figura 3. Localización cromosómica de FUS
Transcritos:
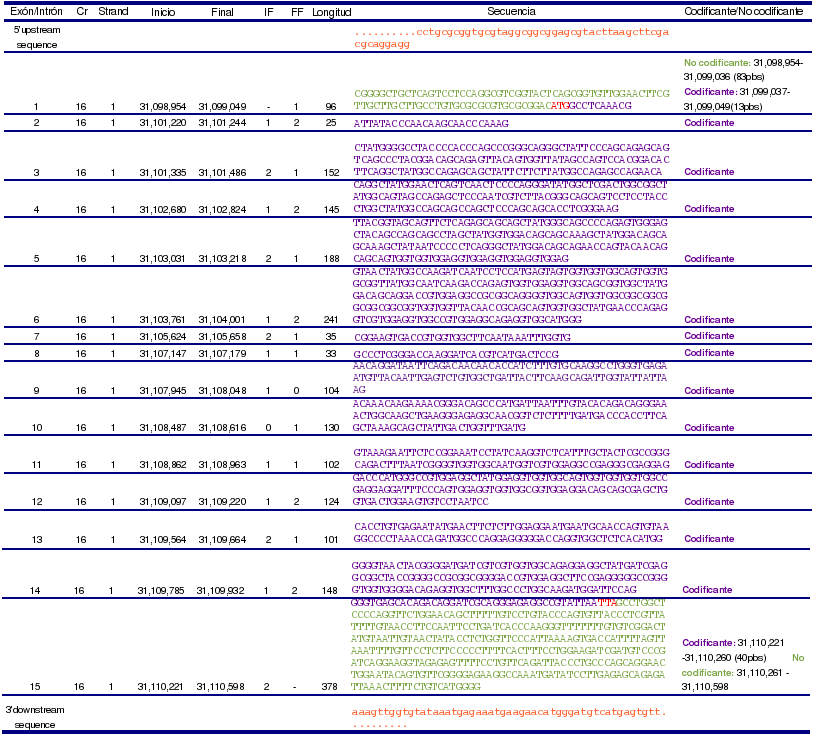
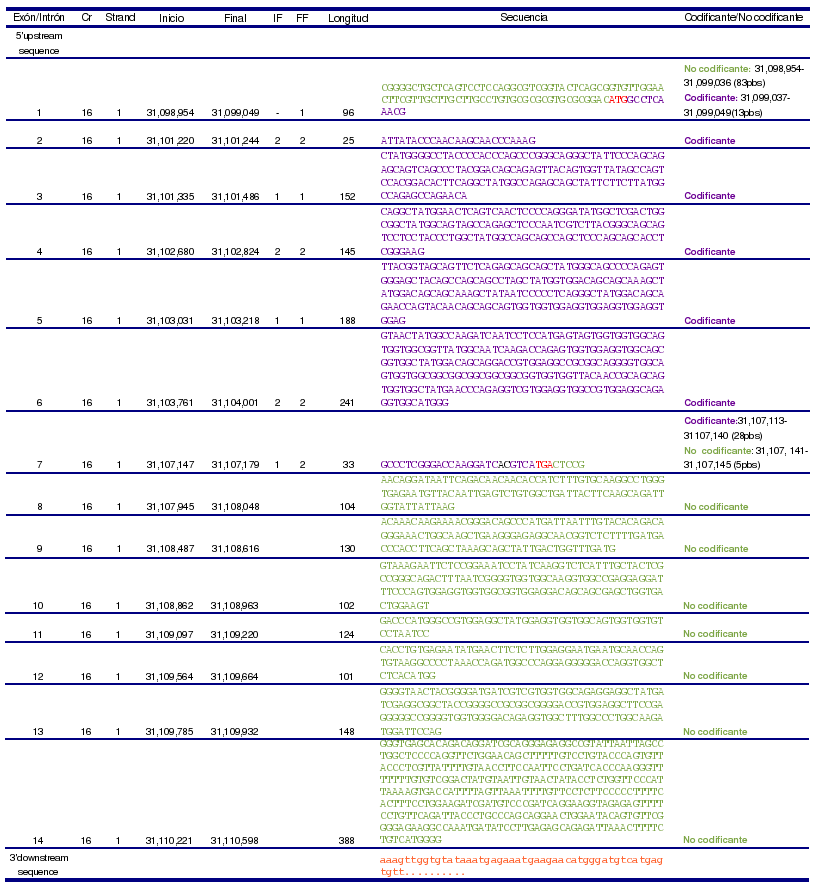
Por medio de splicing alternativo da lugar a 3 transcritos. Las características de cada uno se pueden observar en la siguiente tabla, pero hay que tener en cuenta que la información disponible es diferente según la base de datos que se consulte.
| Transcrito 1 | Transcrito 2 | Transcrito 3 | |
| ID | ENST00000254108 | NM_001010850 | ENST00000380244 |
| Número de exones | 15 | 14 | 14 |
| Longitud transcrito | 2,002 pbs | 1,977 pbs | 1898 pbs |
| Longitud proteína | 526 residuos | 263 residuos | 151 residuos |
| Isoforma | Isoforma A (variante larga y variante corta) | Isoforma B | Isoforma C |
Tabla 1. Información de los tres transcritos de FUS
El transcrito 1 contiene 15 exones y se encuentra asociado con la isoforma a. Ésta tiene dos variantes, conocidas como la variante larga y la variante corta. La variante larga está formada por 526 aminoácidos, mientras que la corta tiene 525 aminoácidos. La diferencia entre ambas se encuentra en que el aminoácido en posición 64 de la variante larga, que es una treonina (T), pasa a ser una serina (S) en la isoforma corta, y que el aminoácido de la posición 65, que es una glicina (G), se pierde. Éstas dos variantes están producidas por splicing alternativo que involucra al exón 4.
Por otro lado, el transcrito 2 contiene 14 exones, y éste se encuentra asociado a la isoforma b. Ésta isoforma está formada por 263 aminoácidos.
La diferencia entre ambos transcritos reside en que, debido al splicing alternativo, el transcrito 2 no tiene el exón que correspondería al exón 7 del transcrito 1. Se produce un cambio en la pauta de lectura en el exón 7 del transcrito 2, que da lugar a la aparición de un codón stop. Esto provoca que en este transcrito, a diferencia del transcrito 1, no todos los exones sean codificantes, y que por este motivo, la proteína codificada por el transcrito 2 sea más corta (263 aminoácidos), que la codificada por el transcrito 1 (526 aminoácidos).
Por último, con respecto al transcrito 3, la información sobre la isoforma C de FUS, no está reconocida en las distintas bases de datos, por tanto no será estudiado en detalle. Este transcrito contiene 14 exones y dará lugar a una proteína de 151 residuos.
Esta proteína tiene gran similitud con la proteína EWS, que es el producto de un gen que se encuentra con mucha frecuencia translocado en el sarcoma de Ewing.
Función: Se une a cadenas sDNA y dsDNA, y promueve la unión de cadenas complementarias, de forma independiente a ATP.
Familia: pertenece a la familia RNP TET.
Subunidad: Componente de los complejos riboproteícos.
Localización subcelular: Nuclear.
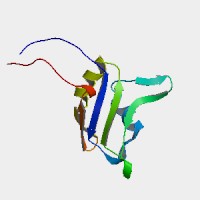
Figura 3. Estructura tridimensional de la proteína FUS
La proteína de fusión FUS-CHOP está producida por una translocación t(12;16)(q13:p11) y es una proteína característica del liposarcoma mixoide. De forma invariable, la primera parte de la proteína de fusión corresponde a la proteína FUS y la segunda a CHOP.
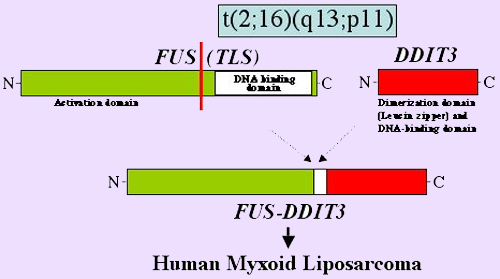
Figura 4. Translocación cromosómica t(12;16)(q13:p11).
La fusión de estas proteínas puede darse de distintas maneras, pero existen tres formas comunes de fusión descritas en la literatura, que dependen de la presencia o ausencia de los exones 6-8 de FUS en el producto de la fusión. El tipo I (también conocido como tipo 7-2) consiste en los exones 1-7 de FUS fusionados con el exón 2 de CHOP. El tipo II (también llamado de tipo 5-2), consiste en la fusión de los exones del 1 al 5 de FUS con el exón 2 de CHOP, y el último tipo, tipo III (o tipo 8-2), que incluye los exones del 1 al 8 de FUS. La fusión de tipo I está involucrada en un 20% de los casos de liposarcoma mixoide, la de tipo II en un 67% y la de tipo III en un 10%.
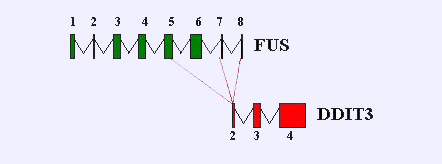
Figura 5. Tipos de fusión entre FUS y CHOP.
En el caso de la proteína que analizamos, la unión tiene lugar entre el exón 7 de FUS y el exón 2 de CHOP, por lo tanto se trata de una proteína de fusión de tipo I.
La proteína resultante de la fusión de los genes FUS-CHOP está formada por 462 aminoácidos, de los cuales los primeros 266 corresponden a FUS. Al producirse la translocación FUS no se transcribe completamente. En cambio, se observa que la transcripción de CHOP si es completa, y que la proteína de fusión contiene los 169 aminoácidos correspondientes. Se puede ver que entre los aminoácidos correspondientes a FUS y los correspondientes a CHOP hay 10 aminoácidos que no corresponden a ninguna de las dos proteínas cuando se transcriben por separado. Estos 10 aminoácidos, provienen de la transcripción de parte del exón 2 de CHOP que no era codificante. Al estar fusionado al gen FUS, se vuelve codificante, por tanto, para analizar CHOP en la proteína de fusión, se usarán los datos del NCBI que corresponden a la proteína CHOP fusionada (CAA63088). A continuación, se muestra el resultado de la fusión.
 |
Se observa, que en el lugar de unión de ambas proteínas aparece una valina. Ésta proviene del último nucleótido del exón 7 de FUS y de los dos primeros del exón 2 de CHOP. Como se trata de 3 nucleótidos, no se producirá un cambio en la pauta de lectura, sino que simplemente, se codifica para un aminoácido nuevo, que no corresponde a ninguna de las dos proteínas, como se puede observar a continuación.
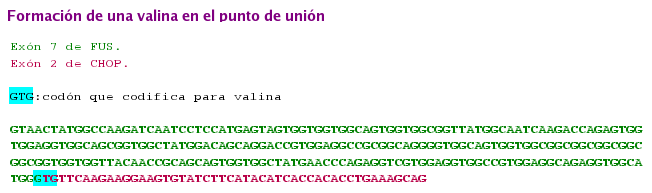 |
Dominios de la proteína FUS-CHOP:
La proteína de fusión contiene los siguientes dominios:
 |
Sabiendo las funciones de las proteínas FUS y CHOP, se puede comprender el papel de la proteína de fusión en el liposarcoma mixoide. CHOP es una proteína nuclear que se encuentra involucrada en la adipogénesis y en la eritropoyesis. Su función consiste en el control del ciclo celular, parando la síntesis de DNA cuando está dañado, gracias al dominio leucine zipper. FUS participa en la integridad genómica, y se une al DNA por medio de los dedos de zinc.
En la proteína de fusión FUS-CHOP, el dominio de unión al RNA de FUS, es reemplazado por la totalidad de la proteína CHOP (dominio de unión a DNA y dominio leucine zipper). Por tanto, FUS es capaz de unirse al DNA, y actúa como un factor de transcripción. CHOP no puede actuar de forma correcta, y hay una desregulación de su función como inhibidor de la transcripción cuando hay daño celular, por lo que las células no pueden parar su proliferación. Es decir, la proteína de fusión actúa como un factor de transcripción anormal, que altera el control de varios genes diana. Por ello, se comprende que la proteína de fusión esté presente en el liposarcoma mixoide.
En la tabla 2 se muestra la conservación del gen FUS en otras especies. Se observa una mayor homología con P. troglodytes, M. musculus y R. norvergicus. Este hecho era previsible ya que se trata del grupo de mamíferos, y filogenéticamente se encuentran más próximos. Al alinear el DNA genómico del ges FUS de H. Sapiens con el de P. troglodites se puede observar que la identidad es del 99%, debido a la proximidad de estas especies. Con los genes ortólogos de las especies más alejadas hay menos conservación. Se puede observar en la tabla que con C. elegans y S.cerevisiae la homología es inferior al 50%.
Tabla 2. Conservación de FUS en otras especies. [% id.= % identidad].
La tabla 3 corresponde a la conservación de la proteína CHOP en otras especies. Las especies que presentan mayor homología con el gen CHOP de H.sapiens son P.troglodytes, M.mulatta y C.familiaris, es decir, el grupo de los mamíferos. La homología en las especies de invertebrados, D.melanogaster y C.elegans, con respecto al gen CHOP del humano es mucho menor.
Tabla 3. Conservación de CHOP en otras especies.[% id.= % identidad].
Para estudiar la expresión de los genes FUS y CHOP en distintos tejidos, nos hemos basado en los datos obtenidos de los resultados de los microarrays obtenidos de la base de datos de UCSC. El resultado se muestra en una tabla en una escala de colores que indican si el gen está sobreexpresado en un tejido, o bien, si su expresión es menor.
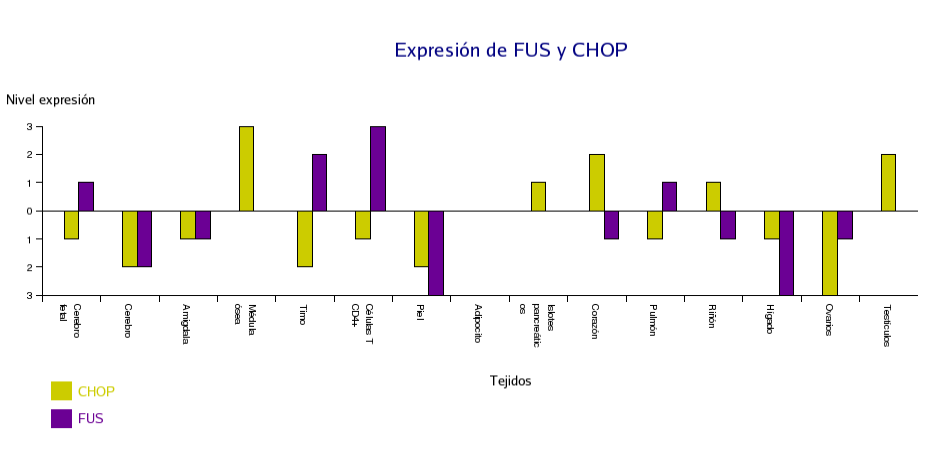
En lo que respecta a la expresión en tejidos, se observa una expresión diferencial de FUS y de CHOP. Los tejidos dónde la expresión de FUS es mayor son el cerebro fetal, el timo, el pulmón y las células T CD4+, sin embargo los tejidos dónde la expresión de CHOP es más elevada son la médula ósea, los islotes pancreáticos, el corazón, el riñón y los testículos. Observamos que FUS y CHOP no se sobreexpresan en los adipocitos. Si se estudiara la expresión de la proteína de fusión, probablemente, se vería una sobreexpresión en este tejido, debido al papel que juega en el liposarcoma mixoide.
|
|
Tabla 4. Expresión de FUS y CHOP en tejidos.
Hemos hecho un gráfico para poder comparar visualmente la expresión diferencial de estos genes en los distintos tejidos. Con el fin de comparar cuantitativamente, tomamos el valor 0 como referencia de la expresión normal en tejido y posteriormente, asignamos valores de -3 a 3 para cuantificar la expresión, desde muy baja (-3) a muy elevada (3).
Con el objetivo de caracterizar la región promotora de los genes FUS y CHOP, hemos creado un programa en lenguaje Perl que permite identificar computacionalmente sitios de unión de factores de transcripción en esta región. Posteriormente, se han comparado con los resultados obtenidos al utilizar como fuente de información el servidor web PROMO. En las tablas 5 y 6, se muestra la lista de los factores de transcripción obtenidos con el programa, la puntuación máxima y la posición de la región promotora a la cual se une el factor de transcripción. Además se muestra el p-value obtenido para cada factor de transcripción, que corresponde a una aproximación a la probabilidad de rechazar erróneamente la hipótesis inicial de que, un factor de transcripción determinado no se une a la secuencia promotora. Además del p-value obtenido con el programa de Perl, también se muestra el p-value equivalente obtenido con el PROMO (ER) de cada uno de los factores de transcripción.
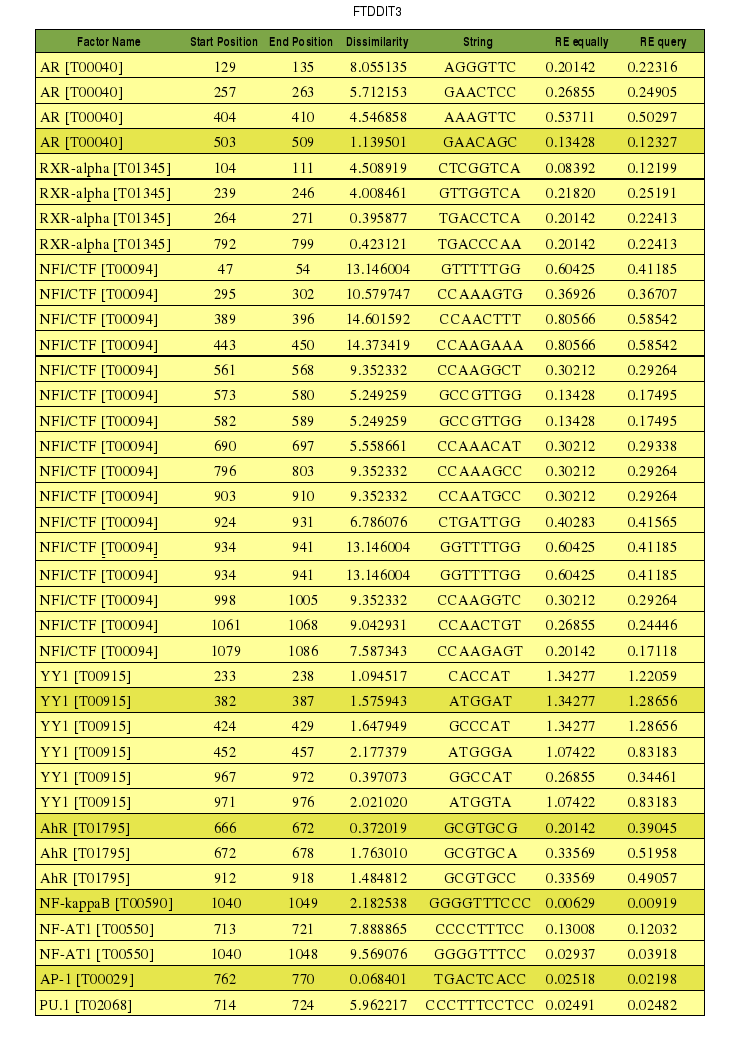
Tabla 5. Promotor del gen CHOP. Comparación del valor p-value obtenido con el programa y con PROMO 1.[Ver resultado programa]
Al observar los resultados obtenidos con el programa en lenguaje Perl para el promotor del gen CHOP ( Tabla 5 ), vemos que sólo un factor de transcripción, el NF-kappaB, tiene un valor de p-value inferior a 0,1. Por tanto, éste factor de transcripción se unirá a la región promotora de CHOP, y será un regulador de su expresión. El resto de factores de transcripción, cuya unión al promotor predice el programa, tienen valores de p-value muy superiores a 0,1 y, en muchos casos, el valor es 1. En consecuencia, en los casos en que obtenemos un valor p-value de 1, podemos confirmar la hipótesis inicial de que este factor de transcripción no se une al promotor y si lo hace, es debido al azar. Al analizar los resultados obtenidos con el PROMO, se observan dos factores de transcripción con un p-value inferior a 0,1. Uno de estos factores es el NF-KappaB, que ya lo mostraba el programa, lo que permite confirmar que este factor de transcripción se une al promotor de CHOP; sin embargo, vemos que los valores de p-value son diferentes, siendo el del PROMO menor. El otro factor de transcripción con un p-value inferior a 0,1 es AP-1, para el cual el programa nos daba un p-value de 1, por tanto este factor según el programa no se unía a la región promotora. El resto de factores de transcripción que predice el PROMO tienen valores de p-value superiores a 0,1. Por otro lado, se observa que con respecto a la posición en la que se une NF-KappaB, mientras que el programa muestra que es la 1041, el PROMO muestra que es la 1040. (Ver tabla de unión de factores de transcripción de CHOP).
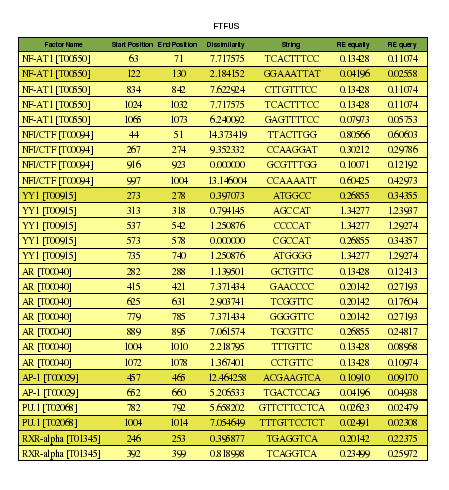
Tabla 6. Promotor del gen FUS. Comparación del valor p-value obtenido con el programa y con PROMO1.[Ver resultado programa]
En este caso, observamos en la tabla 6 los resultados obtenidos con el programa para predecir los factores de transcripción que se unen a la región promotora de FUS, y vemos que no hay ninguno con un p-value inferior a 0,1, sino que en todos los casos éste valor es superior a 0,15. En los casos en los que el valor p-value es de 1, se confirma la hipótesis inicial de que este factor no se une al promotor del gen estudiado y que, si lo hace, es debido al azar. Sin embargo, según el resultado del PROMO, se observan dos factores de transcripción con un p-value inferior a 0,1, estos son el NF-AT1 y el AP-1. El resto de factores de transcripción que predice PROMO tienen un p-value con valores superiores a 0,1. Además, se observan diferencias en la posición a la cual se unen los factores de transcripción dependiendo de si lo hemos predicho con el programa o con el PROMO. (Ver tabla de unión de factores de transcripción de FUS)
Las diferencias que se observan entre lo predicho por el programa que hemos creado, y el PROMO, sobre la unión de diferentes factores de transcripción a los promotores de FUS y de CHOP, son debidas a que el programa que hemos creado se ejecuta con un archivo que contiene solo 13 factores de transcripción, mientras que el PROMO predice multitud de factores de transcripción que se unen a las secuencias promotoras de los genes de interés. Al comparar la posición en la cual se unen los factores de transcripción al promotor, también se observan diferencias, tanto para el gen FUS como para el gen CHOP. Éstas diferencias se deben a que en el programa en lenguaje Perl se usan matrices de 9 elementos en cada fila, mientras que las del PROMO tienen 7.
En la proteína de fusión, CHOP queda bajo el control del promotor de FUS. La proteína contiene la parte 5' terminal de FUS que tiene la función de activar la transcripción. Por tanto, serán los factores de transcripción capaces de unirse a éste promotor, los que regulen la expresión del gen híbrido FUS-CHOP.
Según el análisis con el PROMO, los factores de transcripción que regularán la expresión del gen híbrido FUS-CHOP serán NF-AT1, AP-1 y PU.1.
Para estudiar la función de los genes y ver a qué procesos biológicos están asociados, hemos utilizado la base de datos de Gene Ontology. A continuación se muestran las tablas con las diferentes funciones a las que están asociadas las proteínas FUS y CHOP.
Según la base de datos de Gene Ontology el gen CHOP está asociado a los siguientes GO:
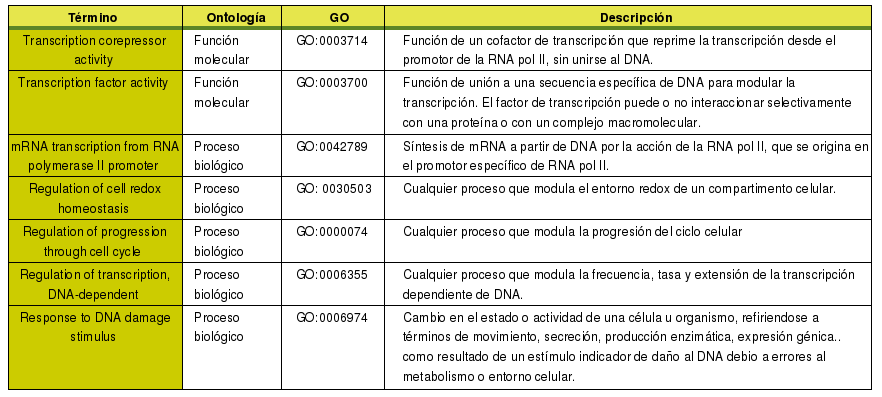
Tabla 7. Término, ontología y descripción del GO asociado a CHOP 1.
En Gene Ontology el gen FUS está asociado a los siguientes términos.

Tabla 8. Término, ontología y descripción del GO asociado a FUS 1.
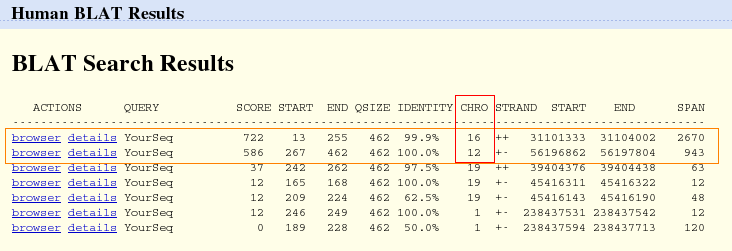
El primer paso al comenzar este trabajo, fue averiguar a que proteína corresponde la secuencia de aminoácidos que nos asignaron bajo el nombre de proteína 4. Para ello, comenzamos haciendo un BLAT desde la base de datos UCSC, con el fin de obtener la parte del genoma que alineaba con esta proteína. Los resultados obtenidos mostraban que, en primer lugar, con un score de 722 y una homología del 99,9% la proteína estaba situada en el cromosoma 16, y se trataba de la proteína FUS. En segundo lugar, con un score de 526 y una homología del 100% se encontraba la proteína CHOP, cuyo gen está situado en el cromosoma 12.
Después de observar los alineamientos con la proteína 4, vimos que la primera región de ésta proteína, según esta base de datos, no alineaba con ninguna parte del genoma. Por tanto, para corroborar los resultados, decidimos hacer un BLAST proteína-proteína (blastp) desde la base de datos NCBI. El resultado obtenido fue que correspondía a la proteína de fusión FUS-CHOP, producto de la translocación entre una región del cromosoma 16 y una región del cromosoma 12, y que está involucrada en el liposarcoma mixoide humano. El score de este resultado es de 390 bits (1003) con una homología del 100% y un E-value de 7e-107.
La información sobre los transcritos del gen FUS es distinta según la base de datos que se utilice para hacer el estudio. En la base de datos del UCSC se mostraban tres transcritos muy similares entre si, y por ello decidimos realizar un alineamiento del mRNA de los transcritos con ClustalW, observando que prácticamente solo variaban en longitud por unos pocos nucleótidos. Por tanto, decidimos descartar el UCSC como base de datos para extraer la información. Por otro lado, en el Ensembl los tres transcritos que se mostraban eran bastante diferentes y en el NCBI solo hay almacenada información sobre dos transcritos, pero, parecía que la información sobre los dos transcritos del NCBI coincidía con la del Ensembl. Partiendo de aquí, decidimos realizar la caracterización de los transcritos de FUS, apoyándonos en la información procedente del NCBI y del Ensembl.
La información sobre el transcrito 1 procedente de ambas bases de datos, es la misma. Pero, al comenzar a analizar el transcrito 2 del gen FUS, vimos que en las bases de datos de NCBI y de Ensembl la información disponible era distinta. En la base de datos del Ensembl este transcrito contiene 13 exones y 1546pbs, mientras que en la del NCBI contiene 14 exones y tiene 1977 pbs. Al alinear ambos transcritos entre sí, se observa que al transcrito que procede del Ensembl le falta el exón 1 casi en su totalidad, y parte del último exón. Además la secuencia de nucleótidos del transcrito del Ensembl comienza con el codón TTT (no con ATG) que codifica para una fenilalanina. Por tanto, consideramos que puede tratarse de un error, y que la secuencia está incompleta. Por estos motivos, escogimos como válido el transcrito 2 de la base de datos del NCBI. El transcrito 3 solo aparece en la base de datos del Ensembl, y por tanto, de allí hemos extraído toda la información.
[Vuelve a transcritos de FUS]Para caracterizar la expresión de los genes y analizar en que tipo de tejidos o células se expresan, hemos usado el programa Gene Sorter, al cual hemos accedido a través de la base de datos del UCSC.
Para estudiar la caracterización de la región promotora de nuestros genes y ver que conjunto de factores de transcripción se unen a esta región promotora hemos:
creado un programa Perl al cual le dábamos un fichero con matrices de factores de transcripción y otro fichero con la secuencia de la región promotora de nuestros genes en formato FASTA. El programa nos mostraba una lista de los factores de transcripción, que contenía el fichero de matrices, que se unían a la secuencia promotora de FUS y CHOP con un valor p-value asociado.
El programa se ha desarrollado en cuatro partes. Una primera parte, donde el programa leía un fichero de matrices de ocurrencias de factores de transcripción y la guardaba en la memoria como un hash de vectores. La segunda parte del programa, consistía en transformar cada una de las matrices de ocurrencias en matrices de pesos, de manera que el resultado mostraba el peso del nucleótido asociado a cada una de las distintas posiciones del motivo. En la tercera parte, se trataba de calcular la puntuación que proporciona cada matriz de pesos a cada posible posición de unión del factor de transcripción dentro de la secuencia promotora, el resultado obtenido mostraba la máxima puntuación y la posición del sitio de unión. En la última parte, el resultado de ejecutar el programa mostraba un valor p-value asociado a cada factor de transcripción. Este resultado era una aproximación a la probabilidad de rechazar erróneamente la hipótesis inicial de que el factor no se una a la región promotora, y por tanto, que si se une sea debido al azar. Por tanto, un valor de p-value cercano a 1 confirma la hipótesis inical de que el factor no se une a nuestra secuencia promotora, mientras que, valores inferiores a 0,1 permiten rechazar la hipótesis inicial.
[Descarga el programa]obtenido el conjunto de factores de transcripción que se unían a nuestra región promotora mediante el servidor web del programa PROMO. Para obtener el listado de factores de transcripción que se unían, usamos un 15% de disimilaridad, y el resultado obtenido mostraba una multitud de factores de transcripción, que era muy difícil de analizar debido a la complejidad de resultados. Por eso, decidimos seleccionar solo los factores de transcripción que estaban en el fichero de matrices, ya que son los que nos interesa comparar, y son los que se utilizan en el programa. Por eso, el resultado del programa PROMO solo muestra los 13 factores de transcripción que corresponden con los que contiene el fichero de matrices.
Para extraer la región promotora de los genes DDIT3 y FUS, hemos usado la base de datos de NCBI, con las características de 1kb upstream y 100bp downstream del TSS (transcription start site).
Para estudiar la función de los genes FUS y CHOP y ver a qué procesos biológicos están asociados, hemos utilizado la base de datos de Gene Ontology.
Se ha estudiado la proteína de fusión FUS-CHOP, que proviene de un gen híbrido formado como consecuencia de una translocación cromosómica, y que se encuentra en la mayoría de los casos de liposarcoma mixoide. Este estudio ha sido complicado de realizar, ya que la información es distinta en las diferentes bases de datos consultadas. Además, al tratarse de una proteína de fusión, ha sido necesario analizar los genes por separado.
Se ha observado que ambos genes están conservados a lo largo de la evolución. La proteína FUS tiene un dominio de unión a RNA, y está involucrada en procesos para mantener la integridad genómica. Por otro lado, la proteína CHOP contiene un dominio leucine zipper, que está relacionado con la regulación de la síntesis de DNA en procesos de daño celular. Se observa que ambos genes están expresados en numerosos tejidos dónde tienen distintas funciones.
La fusión puede tener lugar a través de diferentes exones, dando lugar a distintos tipos de proteína de fusión. En este caso, se ha estudiado la fusión de tipo I, en la cual se une el exón 7 de FUS (transcrito 1) con el exón 2 de CHOP.
Ésta fusión da lugar a una proteína con una nueva función, ya que el dominio de unión al RNA de FUS se pierde, y en su lugar se encuentra la proteína CHOP completa. Ésta proteína actúa como un factor de transcripción, que altera el control de muchos genes diana de CHOP.
Por último, destacar que los factores de transcripción capaces de regular la expresión del gen, son los que son capaces de unirse al promotor de FUS, ya que CHOP está bajo su control en la proteína de fusión.
Páginas web:
http://www.labmed.gu.se/patologi/tumor_biology/myxoid_liposarcoma.htm"
DNA damage-inducible transcript 3; DDIT3 [OMIM id=126337] [disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=126337]
FUSION, derived from 12-16 translocation, malignant liposarcoma; FUS [OMIM id=137070] [disponible en:http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=137070]
Bases de datos y software:
Atlas of Genetics and Cytogenetics in Oncology and Haematology







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}