Raquel Pinacho (raquel.pinacho01@campus.upf.edu) y Federica Eduati (federica.eduati01@campus.upf.edu)
Facultat de Ciències de la Salut i de la Vida
Universitat Pompeu Fabra
La proteína de fusión AML1-MTG8 se forma como consecuencia de la translocación t(8;21)(q22;q22). Mediante esta translocación se produce la fusión de dos genes: AML1 y MTG8. AML1 es un gen implicado en procesos de hematopoyesis, por lo que al formar parte de esta nueva proteína de fusión le confiere ciertas propiedades, que sumadas a las propiedades que obtiene por parte del otro gen implicado en la fusión: MTG8, acaban causando su implicación en el proceso neoplásico que da lugar a la leucemia mieloide aguda.
A partir del estudio de las características de los genes fusionados a consecuencia de dicha translocación, exponemos las características que determinan la propia función de la proteína de fusión AML1-MTG8.
La translocación t(8;21)(q22;q22) es una de las más frecuentes anomalías cromosómicas en la AML (Leucemia mieloide aguda), concretamente, está asociada al subtipo FAB-M2 de AML. Las células leucémicas con la translocación t(8;21)(q22;q22) se caracterizan únicamente por una alta frecuencia de aparición de bastones de Auer y por la maduración de la línea granulocítica. Citogenéticamente, esta translocación suele ir acompañada de la pérdida de cromosomas sexuales, lo que raramente sucede en la AML causada por otros reorganizamientos génicos.
Esta translocación da lugar al reordenamiento de los segmentos génicos procedentes de cada uno de los dos cromosomas (8 y 21), fusionando dos genes AML1 y MTG8, que son los que dan lugar a la proteína de fusión objeto de este estudio: la proteína quimérica AML1-MTG8.
Se ha visto que la translocación interrumpe el cromosoma 21 a la altura de un intrón específico de AML1. Este gen es muy similar al gen runt de segmentación de Drosophila, que codifica un gen nuclear, implicado en la regulación de otros pair-rule genes.
En los pacientes con AML que presentan esta translocación, también es frecuente la presencia del cromosoma 8 derivativo, que se produce como consecuencia de la translocación. Este cromosoma derivativo, suele tener inserido el gen AML1 justo en el punto en que se produce la rotura del cromosoma 8. Así, parece ser que la translocación t(8;21)(q22;q22), hace que la porción 5' del gen AML1 del cromosoma 21 se yuxtaponga en la zona de rotura del cromosoma 8 derivativo, formando un transcrito de fusión. El gen al cual queda unido AML1 es MTG8.
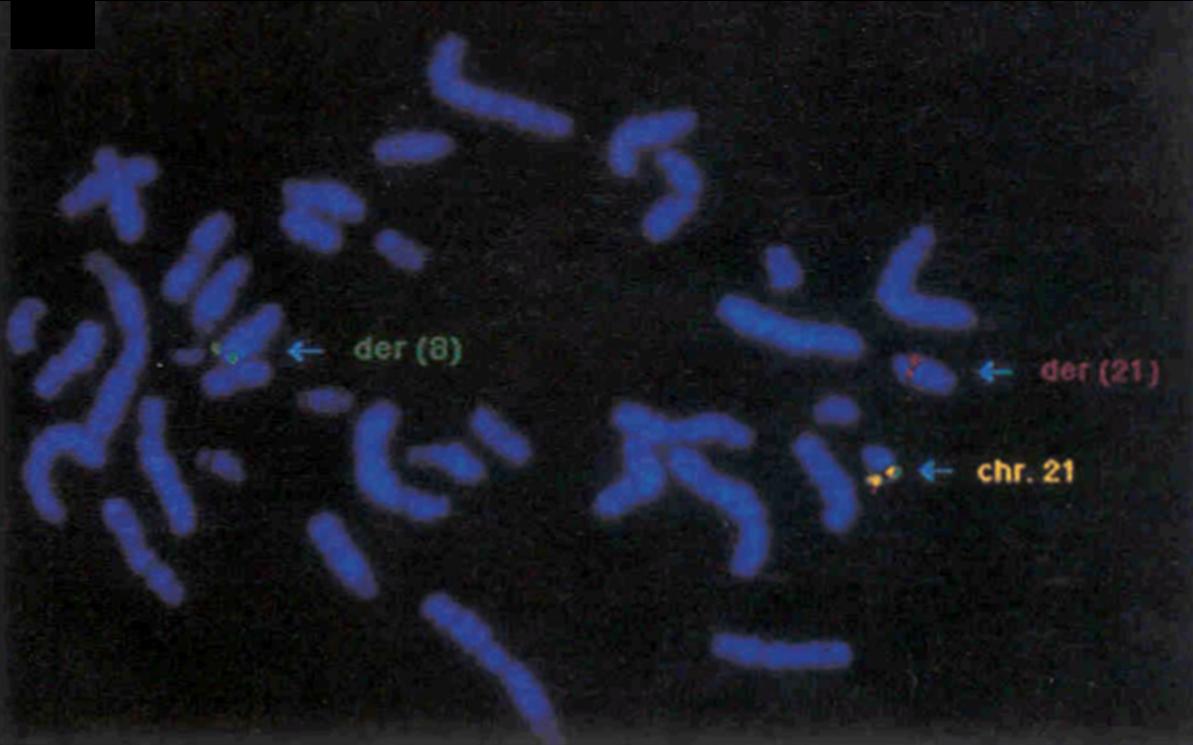
Figura 1: Hibridación in situ, muestra los cromosomas implicados en la translocación t(8;21).
En base a sus características estructurales, se ha inferido que los productos de ambos genes son factores de transcripción.
La expresión del transcrito de fusión AML1-MTG8 es una importante característica de la translocación t(8;21), además la producción de la proteína quimérica está probablemente involucrada de forma directa en la patogénesis de la leucemia mieloide aguda (AML).
A lo largo de este estudio caracterizaremos los genes que dan lugar a dicha proteína de fusión, para tratar de inferir sus propias características génicas, evolutivas, funcionales y de regulación.
En la realización de este estudio, se han aplicado los conocimientos adquiridos a lo largo de la asignatura de Bioinformática. Para facilitar la explicación de las diferentes herramientas utilizadas en la resolución de los apartados propuestos, dividiremos esta sección en los mismos apartados.
En primer lugar se nos pasó una secuencia proteíca desconocida, para su identificación realizamos un blat contra la base de datos de UCSC. Inicialmente, probamos con varias bases de datos, para lo cual también probamos con un blastp en la página del NCBI, ya que dudábamos sobre qué resultados eran más fiables. Al comparar los datos vimos que aunque los nombres variaran, era la misma proteína. En NCBI encontramos la secuencia proteíca de AML1-MTG8 y descubrimos que se trababa de una proteína de fusión producida por una translocación t(8;21) e implicada en un proceso leuquemogénico. Por otra parte, mediante el blat en UCSC, encontramos los genes implicados en la formación del trásncrito de fusión.
Este paso lo resolvimos basándonos principalmente en los datos obtenidos en Ensembl, también fuimos contrastando información con la base de datos de UCSC y la de Uniprot/Swissprot, pero en general nos apoyamos más en lo que encontramos en Ensembl. Para localizar los genes buscamos su identificación en Ensembl, simplemente mediante sus propias herramientas de búsqueda. Los diferentes datos sobre tráscritos, exones y similares se obtienen navegando por la página correspondiente al gen buscado. Los diferentes enlaces que conducen a algunas de las páginas consultadas se encuentran inseridos en el texto correspondiente al apartado Caracterización de la estructura genómica .
En este apartado, el objetivo era obtener información sobre las posibles especies que conservaran genes ortólogos a los genes de interés. Un gen ortólogo es un gen que comparte un origen común con nuestro gen, pero además realiza una función similar. Para estudiar los ortólogos potenciales de ambos genes (AML1 y MTG8) consultamos nuevamente la base de datos de Ensembl, ya que contiene datos de ortología bastante fiables. De todos modos, hay que tener en cuenta que únicamente contiene datos sobre aquellas especies cuyo genoma ha sido secuenciado y además muchos de ellos son sólo predicciones (no se ha caracterizado su función con exactitud), por lo que es posible que alguno de los resultados no sea realmente un gen ortólogo, sino simplemente un homólogo, o también es posible que existan ortólogos en otras especies cuyo material genético no haya sido estudiado.
En este apartado, el objetivo era evaluar la expresión de cada uno de los genes. Con este propósito, utilizamos la base de datos de UCSC, donde tienen datos de expresión en forma de microarrays. Dentro de la misma base de datos, consultamos tambíén la aplicación VisiGene, para excontrar imágenes de diferentes preparaciones tisulares que mostrasen la expresión diferencial de los genes. En este apartado, también consultamos diferentes artículos encontrados en el apartado del Pubmed del NCBI, para contrastar un poco la información y que fuera algo más exacta.
Para este apartado fueron necesarias dos aproximaciones en paralelo. Por un lado, mediante un programa realizado por nosotras mismas y una serie de matrices de pesos de 13 factores de transcripción que nos fueron proporcionados, habíamos de determinar cuáles de ellos eran buenos candidatos de interaccionar con la región promotora de nuestros genes (1000 nts upstream del punto de inicio de la transcripción y 100 downstream).
Utilizando la misma secuencia promotora, teníamos que buscar todos los posibles factores de trasncripción humanos que se unieran a ella. Para ello utilizamos la aplicación PROMO. Inicialmente, realizamos este paso con un porcentaje de disimilaridad del 15%, pero como nos salían demasiados factores de transcripción, lo bajamos al 3%, que nos pareció suficiente para obtener resultados más fiables al limitar la búsqueda a factores con menor porcentaje de disimilaridad respecto a la secuencia.
En ambos casos era necesario establecer un umbral apartir del cual considerar un factor de transcripción como válido, decidimos considerar todos aquellos que tuvieran un p-value inferior a 0,09.
Para conocer la función de los genes, consultamos las anotaciones de ontología que había en las diferentes bases de datos, concretamente visitamos Ensemble, Uniprot y la página web de GeneOntology.
Para comletar la información y obtener una idea más exacta de las fuciones de los genes y de nuestra proteína de fusión, consultamos la literatura disponible en Pubmed (NCBI), de la cual hicimos una selección (ver referencias) que nos ha permitido comprender el papel de AML1-MTG8 en la patogénesis de la leucemia mieloide aguda y cómo los diferentes dominios heredados de los genes que se fusionan mediante la translocación, son responsables de la actividad de la proteína de fusión resultante.
AML1-MTG8 es una proteína de fusión, producto de una translocación entre los cromosomas 21 y 8. Por ello, para deducir su estructura genómica, primero estudiamos la estructura genómica de los genes de los que procede. Para averiguar cuáles son, realizamos un blat de nuestra secuencia de proteína contra la base de datos (UCSC). Este blat nos proporcionó una serie de secuencias que tenían altos puntuaciones de similitud. Selecionamos dos de ellas, las que presentaban mayor puntuación de similitud y mayor porcentaje de identidad: las correspondientes a los genes AML1 y MTG8. Estos son los genes que vamos a caraterizar a lo largo de este trabajo.
Una de las entradas que nos mostró el blat nos condujo a AML1 (100% de identidad). En la imagen vemos un resumen de sus principales características:
Figura 2: Estructura general del gen AML1
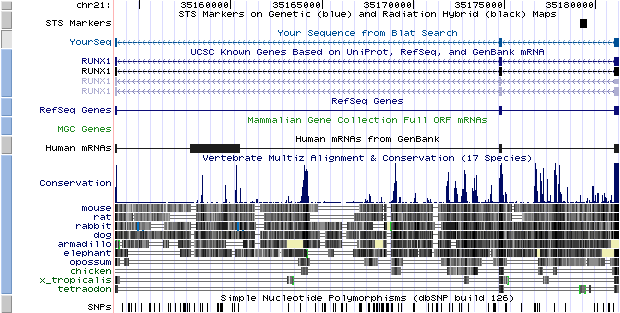
Figura 3: Esquema de los exones que componen el gen AML1.

Como se obsera en la imagen, este gen contiene 9 exones diferentes. Los exones dan lugar a los diferentes tránscritos de AML1 mediante splicing alternativo. Los exones coloreados en negro contienen regiones no traducidas (UTRs), mientras que los exones coloreados en blanco representan exones codificantes. Así podemos clasificar los exones de AML1 según éstos contengan regiones codificantes o no.
Exones con regiones codificantes: exones 2, 4, 5, 6 y 7b completos y parte de los exones 1, 3, 7a y 8.
Exones que contienen regiones no codificantes: porciones de los exones 1, 3, 7a y 8.
Figura 4: Esquema de los diferentes tránscritos que se producen a partir del gen AML1.
![]()
A continuación caracterizamos los diferentes tránscritos que se originan a partir de AML1, a partir de la información encontrada en Ensembl.
Figura 5: Estructura del tránscrito ENST00000300305.
![]()
Su primer exon , (ENSE00001317351), de 503 nucleótidos, tiene los primeros 445 nucleótidos que son no codificantes. También, en el último exón (ENSE00001247313) contiene una región no codificante, que comprende los últimos 4327 nucleótidos del mismo exón. El resto de los exones que lo componen no contienen UTRs. Al observar las figuras de la estructura de los tránscritos hay que tener en cuenta que se trata de la hebra reversa, por lo que el orden de los exones va de derecha a izquierda como indica la flecha.
Figura 6: Estructura del tránscrito ENST00000325074.
![]()
El último exón (ENSE00001247313)de 4803 nucleótidos, tiene 4327 nucleótidos no codificantes. El resto son codificantes. Al observar las figuras de la estructura de los tránscritos hay que tener en cuenta que se trata de la hebra reversa, por lo que el orden de los exones va de derecha a izquierda como indica la flecha.
Figura 7: Estructura del tránscrito ENST00000342083.
![]()
El primer exón (ENSE00001380483)de 1848 nucleótidos, tiene 1578 nucleótidos no codificantes. El resto son codificantes. Al observar las figuras de la estructura de los tránscritos hay que tener en cuenta que se trata de la hebra reversa, por lo que el orden de los exones va de derecha a izquierda como indica la flecha.
Figura 8: Estructura del tránscrito ENST00000344691.
![]()
El primer exón (ENSE00001380483) de 1848 nucleótidos, tiene 1578 nucleótidos no codificantes. El resto de los exones son codificantes. Al observar las figuras de la estructura de los tránscritos hay que tener en cuenta que se trata de la hebra reversa, por lo que el orden de los exones va de derecha a izquierda como indica la flecha.
Figura 9: Estructura del tránscrito ENST00000358356.
![]()
El primer exón (ENSE00001380483) de 1848 nucleótidos, tiene 1578 nucleótidos no codificantes y el último (ENSE00001400245), que tiene 391 nucleótidos no codificantes al final del exon, de un total de 420 nucleótidos tiene 391 nucleótidos no codificantes, el resto de los exones son en su totalidad codificantes. Al observar las figuras de la estructura de los tránscritos hay que tener en cuenta que se trata de la hebra reversa, por lo que el orden de los exones va de derecha a izquierda como indica la flecha.
Al comparar los tránscritos entre sí, vemos que sólo hay dos exones que todos los tránscritos tienen en común: ENSE00001247303 y ENSE00001044058. Los diferentes tránscritos se generan por splicing alternativo, como hemos visto al comparar las proteínas que generan. Comparando las mismas proteínas hemos deducido que tampoco cambia el marco abierto de lectura, ya que las secuencias aminoacídicas son muy semejantes entre sí, como puede verse en este alineamiento múltiple realizado con clustalw.
(Volver a Estructura genómica).La segunda entrada con mayor porcentaje de identidad (99,9%), nos llevó a la proteína MTG8, cuyas características principales son las que vemos en la imagen:
Figura 10: Estructura del gen MTG8.
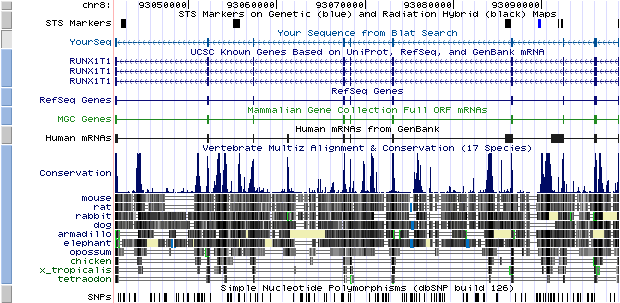
Figura 11: Esquema de los exones que componen el gen MTG8.
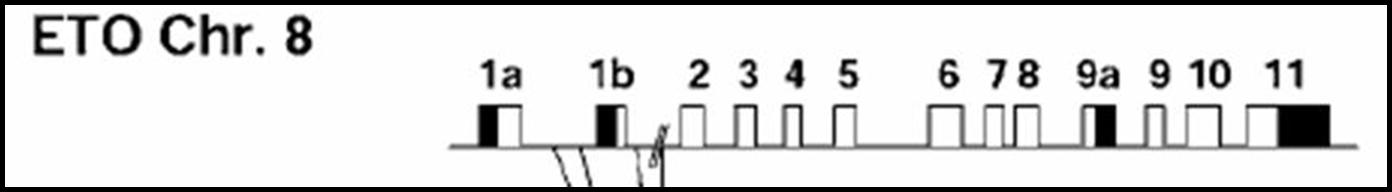
Como se obsera en la figura 5, el gen MTG8 está formado por 13 exones diferentes. Los exones dan lugar a los diferentes tránscritos de MTG8 mediante splicing alternativo. Los exones coloreados en negro contienen regiones no traducidas (UTRs), mientras que los exones coloreados en blanco representan exones codificantes. Así podemos clasificar los exones de MTG8 según éstos contengan regiones codificantes o no.
Exones con regiones codificantes: exones 2, 3, 4, 5, 6, 7, 8, 9 y 10 completos y parte de los exones 1a, 1b, 9a y 11.
Exones que contienen regiones no codificantes: porciones de los exones 1a, 1b, 9a y 11.
Figura 12: Esquema de los tránscritos de MTG8.
![]()
Figura 13: Estructura del tránscrito ENST00000265814.
![]()
En el primer exón (ENSE00001297181), de 172 nucleótidos), los primeros 86 nucleótidos son no codificantes; por otro lado, en el último exon (ENSE00001226380), en este caso de 1594 nucleótidos, los 1342 últimos son no codificantes. El resto de exones son codificantes, es decir, no contienen UTRs.
Nota: la figura muestra la cadena reversa, por lo que el orden de los exones va de derecha a izquierda, es decir, en el sentido de la flecha.
Figura 14: Estructura del tránscrito ENST00000360348.
![]()
El primer exón (ENSE00001423245), de 188 nucleótidos es no codificante. El segundo (ENSE00001372738) de 138 nucleótidos, comienza con 24 nucleótidos no codificantes. En cuanto a el último (ENSE00001226308), tiene en total 1594 nucleótidos, de los cuales los 1318 últimos son no codificantes. El resto de los exones son codificantes.
Nota: la figura muestra la cadena reversa, por lo que el orden de los exones va de derecha a izquierda, es decir, en el sentido de la flecha.
Al comparar los tránscritos entre sí, vemos que sólo en los dos tránscritos los primeros dos exones son diferentes. En el primer tránscrito, el primer exón es el ENSE00001297181 y en el segundo tránscrito es ENSE00001423245; en el primer tránscrito el segundo exon es el ENSE00001168648 y en el segundo tránscrito es el ENSE00001372738. Los exones corresponden a secuencias codificantes, por lo que dan lugar a dos isoformas diferentes (splicing alternativo).
Al alinear ambas isoformas, vemos que el porcentaje de identidad es muy alto, por lo que hemos deducido, que a pesar de sufrir splicing alternativo para alguno de sus exones, éste no afecta al marco abierto de lectura.
En la literatura hemos encontrado que MTG8 da lugar a 4 isoformas por splicing alternativo, y que está regulado por dos promotores. Como en Ensembl únicamente se mencionan dos tránscritos hemos decidido ceñirnos a su criterio, además otras bases de datos (Uniprot/Swissprot) coinciden en que MTG8 da lugar a dos isoformas: MTG8a y MTG8b; por lo tanto, confirma la información obtenida en Ensembl.
Mediante la comparación entre los diferentes tránscritos de ambos genes, hemos deducido, qué exones son los que están implicados en la generación de nuestra proteína de fusión.
| Gen de procedencia | Exones |
|---|---|
| AML1 (residuos 1-177) |
ENSE00001380483 |
| ENSE00001247303 | |
| ENSE00001044058 | |
| MTG8 (residuos 178-752) |
ENSE00001168648 |
| ENSE00000699743 | |
| ENSE00000699741 | |
| ENSE00000699738 | |
| ENSE00000699731 | |
| ENSE00000699729 | |
| ENSE00000699727 | |
| ENSE00001226308 | |
| ENSE00001226380 |
De todos modos, hemos encontrado un esquema en la literatura, que indica específicamente cómo se genera la proteína de fusión, por lo que consideramos interesante ańadirlo aquí.
Figura 15: Generación de la proteína de fusión AML1-MTG8.
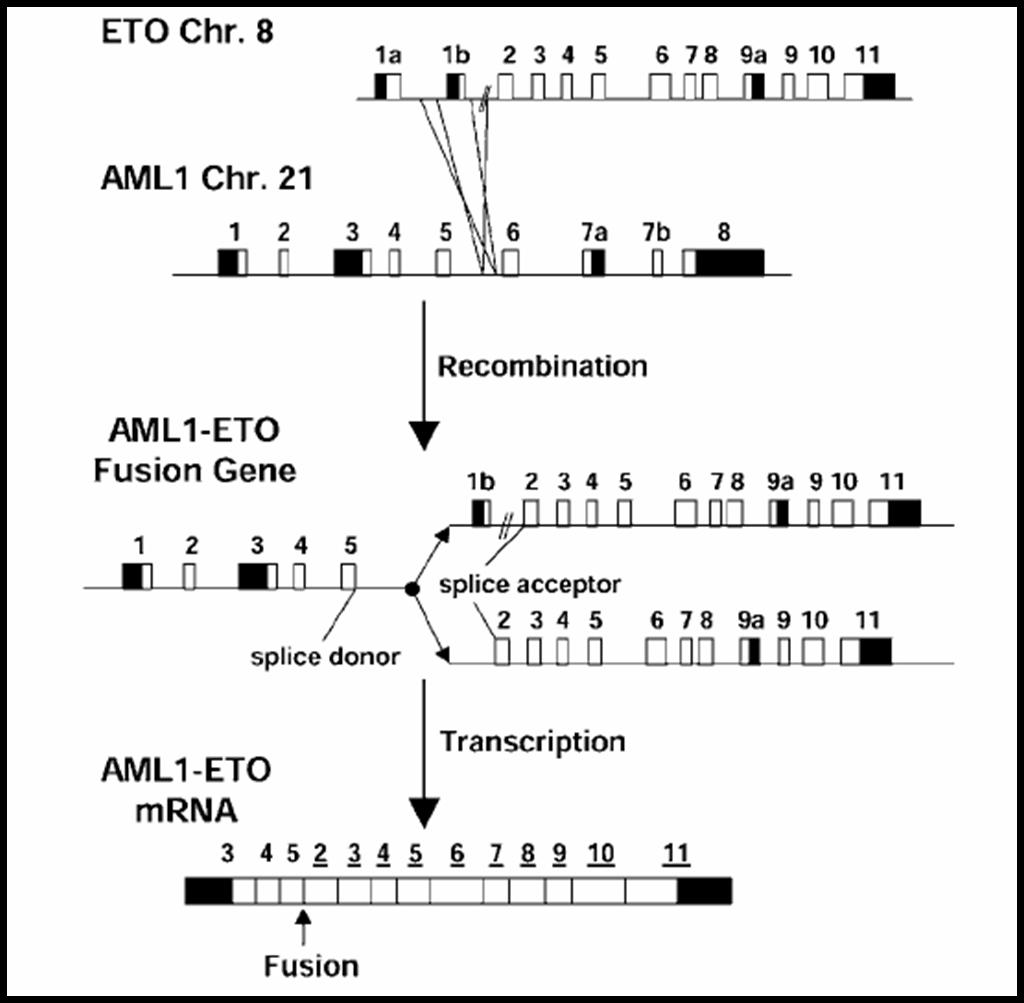
En la figura, se muestran las zonas donde la translocación produce el breakpoint mediante unas líneas transversales. Se observa un splice donor al final del exón 5 de la porción procedente de AML1 (concretamente en el intrón 5, en 3 regiones conocidas como breakpoint cluster regions o BCR), que se unirá a un splice acceptor del exón 2 (en este gen, encontramos 1 BCR en el intrón 1a y 3 en el 1b) de la porción de MTG8a o MTG8b.
El nucleótido 2110 del gen de AML1 es el punto donde la translocación interrumpe el exón, los fragmentos procedentes de ambos genes sufren recombinación lo que dará lugar al transcrito de fusión, en este proceso también median procesos de splicing, como se menciona en el párrafo anterior.
Como curiosidad, mencionar que parece ser que los BCRs de AML1 y MTG8 están asociados con la degradación mediante DNAtopoisomerasa II y sitios hipersensibles a DNAsa I, lo que probablemente esté implicado en la ruptura específica de la cadena de DNA en estas regiones.
Un gen ortólogo es una copia del gen que se encuentra en otra especie, es decir, que comparte un origen común con nuestro gen, pero además realiza una función similar. Para estudiar los ortólogos potenciales de AML1 podemos consultar Ensembl, ya que contiene datos de ortología bastante fiables. Para el gen AML1, las especies que presentan genes ortólogos son las siguientes, destacaremos entre ellas las que tengan un porcentaje de homología por encima del 90% (celdas coloreadas), ya que creemos que serán los ortólogos más cercanos al gen AML1 humano.
| Especie | Tipo de ortología | Gen ID | Porcentaje de homología (%) |
|---|---|---|---|
| Aedes aegypti | many-to-many | AAEL006160 | 21 |
| many-to-many | AAEL006167 | 26 | |
| many-to-many | AAEL007036 | 16 | |
| many-to-many | AAEL007040 | 20 | |
| Anopheles gambiae | many-to-many | ENSANGG00000011627 | 20 |
| many-to-many | ENSANGG00000019555 | 19 | |
| Bos taurus | 1-to-1 | ENSBTAG00000004742 | 98 |
| Caenorhabditis elegans | 1-to-many | B0414.2 | 18 |
| Canis familiaris | 1-to-1 | ENSCAFG00000009596 | 92 |
| Cavia porcellus | 1-to-1 | ENSCPOG00000002684 | 62 |
| Ciona intestinalis | 1-to-many | ENSCING00000002253 | 24 |
| Dasypus novemcinctus | 1-to-1 | ENSDNOG00000015041 | 46 |
| Drosophila melanogaster | many-to-many | CG1379 | 45 |
| many-to-many | CG15455 | 22 | |
| many-to-many | CG1689 | 30 | |
| many-to-many | CG1849 | 27 | |
| Echinops telfairi | 1-to-1 | ENSETEG00000011313 | 86 |
| Felis catus | 1-to-1 | ENSFCAG00000006761 | 65 |
| Gallus gallus | 1-to-1 (aparente) |
ENSGALG00000016022 | 66 |
| Gasterosteus aculeatus | 1-to-1 | ENSGACG00000015276 | 49 |
| Loxodonta africana | 1-to-1 | ENSLAFG00000004290 | 47 |
| Macaca mulatta | 1-to-1 | ENSMMUG00000001649 | 99 |
| Monodelphis domestica | 1-to-1 (aparente) |
ENSMODG00000021051 | 85 |
| Mus musculus | 1-to-1 (aparente) |
ENSMUSG00000022952 | 89 |
| Ornithorhynchus anatinus | 1-to-1 | ENSOANG00000011162 | 12 |
| Oryctolagus cuniculus | 1-to-1 | ENSOCUG00000013731 | 93 |
| Oryzias latipes | 1-to-1 | ENSORLG00000020699 | 55 |
| Pan troglodytes | 1-to-1 | ENSPTRG00000013883 | 100 |
| Rattus norvegicus | 1-to-1 (aparente) | ENSRNOG00000001704 | 88 |
| Takifugu rubripes | 1-to-1 | SINFRUG00000149207 | 57 |
| Tetraodon nigroviridis | 1-to-1 | GSTENG00034595001 | 34 |
| Tupaia belangeri | 1-to-1 | ENSTBEG00000000192 | 74 |
| Xenopus tropicalis | 1-to-1 (aparente) | ENSXETG00000014140 | 82 |
Como hemos visto en la tabla, las especies que presentan genes ortólogos al AML1 humano son: Bos taurus, Canis familiaris, Macaca mulatta, Oryctolagus cuniculus y Pan troglodytes.
Contrariamente al porcentaje de homología que hemos observado en Ensemble para los ortólogos en Drosophila melanogaster (varía entre 27-45%), la literatura cita contínuamente la relación de homología que tienen, ya que comparten un dominio runt de unión a DNA. Así AML1 pertenece a la familia RUNX, por poseer dicho dominio runt de unión a DNA, familia a la que también pertenece el gen runt de la segmentación de Drosophila. Así aunque el porcentaje de homología no sea muy alto, son ortólogos.
Para hallar las especies con genes ortólogos al gen MTG8 humano, hemos seguido el mismo procedimiento que para hallar las de AML1. Las espécies que presentan genes ortólogos son las siguientes, remarcamos en otro color aquellas con homología superior al 90%.
| Especie | Tipo de ortología | Gen ID | Porcentaje de homología (%) |
|---|---|---|---|
| Aedes aegypti | many-to-many | AAEL003615 | 26 |
| many-to-many | AAEL014203 | 26 | |
| Anopheles gambiae | 1-to-many | ENSANGG00000015731 | 26 |
| Bos taurus | 1-to-1 | ENSBTAG00000017339 | 94 |
| Canis familiaris | 1-to-1 | ENSCAFG00000009078 | 92 |
| Danio rerio | 1-to-1 | ENSDARG00000003680 | 87 |
| Dasypus novemcinctus | 1-to-1 (aparente) | ENSDNOG00000008847 | 51 |
| Drosophila melanogaster | 1-to-many | CG3385 | 24 |
| Echinops telfairi | 1-to-1 (aparente) | ENSETEG00000014837 | 75 |
| Erinaceus europaeus | 1-to-1 (aparente) | ENSEEUG00000004013 | 77 |
| Felis catus | 1-to-1 | ENSFCAG00000005350 | 85 |
| Gallus gallus | 1-to-1 (aparente) | ENSGALG00000015926 | 97 |
| Gasterosteus aculeatus | 1-to-1 | ENSGACG00000005059 | 84 |
| Loxodonta africana | 1-to-1 (aparente) | ENSLAFG00000005779 | 84 |
| Macaca mulatta | 1-to-1 (aparente) | ENSMMUG00000023115 | 99 |
| Monodelphis domestica | 1-to-1 (aparente) | ENSMODG00000007500 | 92 |
| Mus musculus | 1-to-1 (aparente) | ENSMUSG00000006586 | 94 |
| Ornithorhynchus anatinus | 1-to-1 (aparente) | ENSOANG00000001913 | 92 |
| Oryctolagus cuniculus | 1-to-1 (aparente) | ENSOCUG00000003754 | 89 |
| Oryzias latipes | 1-to-1 | ENSORLG00000012311 | 80 |
| Pan troglodytes | 1-to-1 | ENSPTRG00000020416 | 100 |
| Rattus norvegicus | 1-to-1 (aparente) | ENSRNOG00000005673 | 94 |
| Takifugu rubripes | 1-to-1 | SINFRUG00000133596 | 80 |
| Tetraodon nigroviridis | 1-to-1 | GSTENG00017458001 | 80 |
| Tupaia belangeri | 1-to-1 | ENSTBEG00000010312 | 98 |
| Xenopus tropicalis | 1-to-1 | ENSXETG00000014592 | 92 |
Como se observa en la tabla, las especies con genes ortólogos que presentan la mayor homología, son: Bos taurus, Canis familiaris, Gallus gallus, Macaca mulatta, Monodelphis domestica, Mus musculus, Ornithorhynchus anatinus, Pan troglodytes, Rattus norvegicus, Tupaia belangeri y Xenopus tropicalis . Como curiosidad, comentar que varias de estas especies son peces, lo que nos ha sorprendido por el alto porcentaje de homología que presentan.
Volver a Conservación en otras especies.
Para la carecterización de la expresión, utilizaremos los diferentes recursos trabajados en clase, como VisiGene y la aplicación de Microarray Expression Data (ambos de UCSC). Contrastando los resultados obtenidos mediante ambas aplicaciones con la información obtenida mediante bibliografía, presentamos las siguientes tablas de la distribución tisular de la expresión de ambos genes.
Este gen se expresa en todos los tejidos examinados, excepto en cerebro y corazón. Presenta los niveles de expresión más elevados en timo, médula ósea y sangre periférica (especialmente en el linaje de linfocitos T). La tabla 1 muestra los niveles de expresión según la Microarray Expression Data de UCSC.
Tabla 1: Patrón de expresión de AML1.
| Tejido | Cerebro | Timo | Sangre y células sanguíneas | Bazo | Adipocitos | Tiroides fetal | Músculo liso | Músculo esquelético | Corazón | Miocitos cardíacos | Pericardio | Páncreas | Próstata | Glándula adrenal | Pulmón | Aparato reproductor | Riñón y aparato urinario |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nivel de expresión | - | +++ | +++ | ++ | + | + | ++ | - | - | + | + | - | - | - | + | + | + |
| Alta | Media | Baja | Nula | |
| Nivel de expresión | +++ | ++ | + | - |
En VisiGene, hemos encontrado diferentes secciones de cerebro de ratón que muestran el patrón de expresión del gen en dicha área. Presentamos a continuación las imágenes que nos han parecido más relevantes.
Figura 16: muestra un embrión de ratón de 11 días. El corte muestra el embrión entero. Se observan en blanco zonas dónde se expresa AML1 de forma activa.
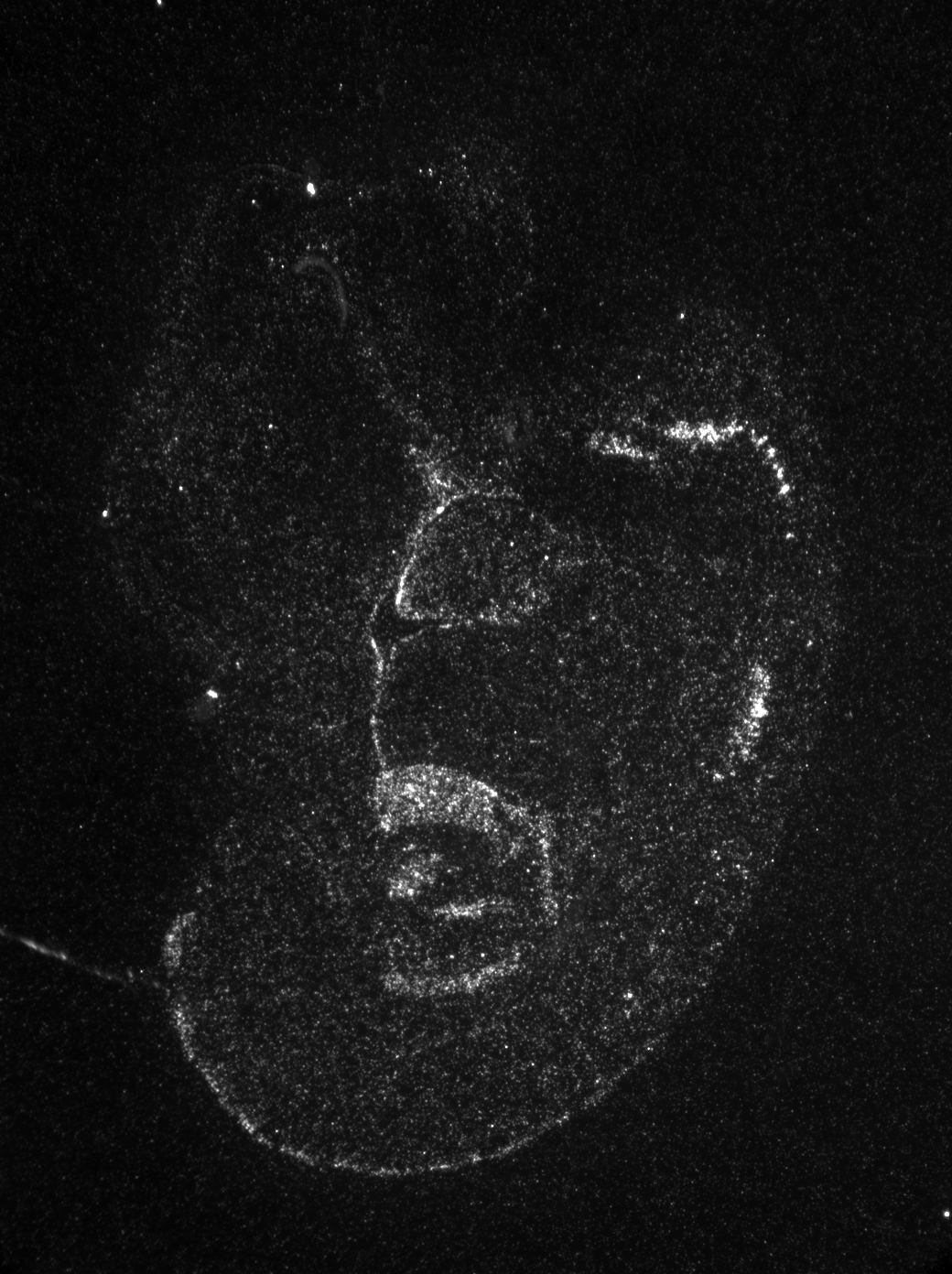
Figura 17: ratón de 7 días de vida. El corte muestra una sección de la médula espinal, se observa que la expresión de AML1 queda restringida al cuerno dorsal.

Según la literatura, no se ha encontrado expresión detallada de AML1 en el Sistema Nervioso Central. Ambas fotografías se han realizado a partir de un experimento en el que se asocia un gen reporter a RUNX1 (lacZ), mediante un Bac. Se observa que el gen lacZ se expresa mayoritariamente en la circulación cerebral, y de forma restringida en médula espinal y MOb (Medulla oblongata). De todos modos, estos datos obtenidos a partir de VisiGene no son concluyentes, ya que en el mismo pie de foto se indica que es necesario corroborarlos mediante un microarray.
En conclusión, AML1 se expresa principalmente en timo, médula ósea y sangre periférica, lo que parece indicar su implicación en el proceso de hematopoyesis, especialmente en la maduración de la línea linfoide, ya que el timo y la médula ósea son los principales órganos donde tiene lugar la maduración de estas células. Esto es un hecho muy importante para poder explicar la implicación de la proteína de fusión AML1-MTG8 en la patogénesis de la leucemia mieloide aguda, como explicaremos de forma detallada más adelante.
Este gen, al contrario de lo que sucede con AML1, se expresa principalmente en el Sistema Nervioso Central (Encéfalo y Médula espinal). Presenta expresión también en otras áreas (pulmones, corazón, testículos y ovarios), aunque en niveles más bajos.
Tabla 2: resumen de los datos obtenidos mediante el miroarray de UCSC.
| Tejido | Cerebro | Riñón | Hígado | Testículo | Ovario | Médula espinal | Ojo | Timo | Bazo | Nodo linfático | Médula ósea | Glándula mamaria | Glándula adrenal | Próstata | Tiroides | Hueso | Tejido adiposo | Grasa marrón | Vejiga | Corazón | Músculo esquelético | Útero | Glándula salival | Estómago | Intestino | Vesícula biliar | Hígado | Riñón | Pulmones | Tráquea | Dedos | Epidermis | Lengua | Placenta | Cordón umbilical |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nivel de expresión | ++ | - | + | - | - | ++ | ++ | - | + | + | + | ++ | ++ | - | - | + | + | + | + | - | + | - | + | + | + | + | + | - | + | + | - | + | - | - | + |
| Alta | Media | Baja | Nula | |
| Nivel de expresión | +++ | ++ | + | - |
En VisiGene, hemos encontrado diferentes secciones de ratón, en las que se puede observar el patrón de expresión del gen MTG8 en diferentes áreas. Presentamos a continuación las imágenes que nos han parecido más relevantes.
Figura 18: Cría de ratón wild type de 7 días de edad. La imagen muestra diferentes cortes del encéfalo (sagital, coronal, transversal). En general, se observa expresión del gen objeto de estudio en aquellas áreas que aparecen más blancas en la imagen.

Figura: 19 Embrión de 11 días. Se muestra un corte transversal del embrión completo. Como puede verse por la intensidad de la luminosidad en ciertas áreas, MTG8 se expresa de forma preferencial en los tejidos que darán lugar al sistema nervioso central. De todos modos, observamos que la expresión de MTG8 es bastante generalizada.
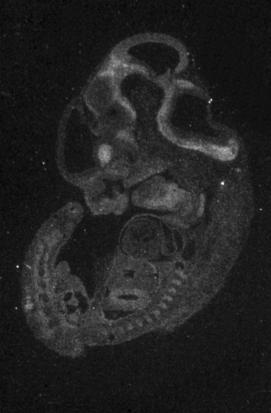
Hemos visto que MTG8 se expresa en órganos distintos a dónde se expresa AML1, lo que nos abre un interrogante: żdónde se expresará la proteína de fusión fruto de ambos genes? De momento no conocemos su distribución tisular, es posible que sea una mezcla de la distribución de los genes que la conforman. Pero, sin duda, ha de expresarse en la línea hematopoyética y probablemente en los órganos donde se produce dicha maduración (timo, médula ósea), ya que sí se conoce su fuerte implicación en el ya mencionado proceso neoplásico. Su contribución a dicho proceso será comentada en el apartado de Caracterización de la función del gen.
Volver a Caracterización de la expresión.
Para caracterizar la región promotora de este gen hemos aplicado dos protocolos diferentes: análisis de la secuencia promotora con un programa propio, para dedicir qué factores de transcripción de los propuestos se unen a nuestra secuencia promotora. La otra aproximación al problema se realiza mediante la aplicación PROMO.
Hemos hecho un programa en perl*, para analizar la secuencia promotora de AML1, a partir de dicho programa, se obtienen unos resultados que mostramos resumidos en la siguiente tabla:
*Visualizar programa en pantalla.
| Nombre matriz | Score | Posición | p-value |
|---|---|---|---|
| AP-1 [T00029] | 4.9195 | 429 | 0.01 |
| AR [T00040] | 0.9595 | 508 | 0.57 |
| c-Myc [T00140] | -993.7085 | 298 | 0.39 |
| NF-AT1 [T00550] | -0.9830 | 500 | 0.33 |
| NF-kappaB [T00590] | -1.7907 | 115 | 0.06 |
| SRF [T00764] | -998.3673 | 69 | 0.31 |
| YY1 [T00915] | 1.1605 | 498 | 0.69 |
| RXR-alpha [T01345] | 1.3616 | 352 | 0.76 |
| HIF-1 [T01609] | -997.0124 | 272 | 0.39 |
| AhR [T01795] | -996.9583 | 534 | 0.91 |
| PU.1 [T02068] | -1.5537 | 464 | 0.32 |
| HNF-4 [T02758] | -996.1412 | 317 | 0.24 |
| NRSF [T06124] | -1994.1732 | 19 | 0.18 |
Marcamos en un color más claro aquellos factores que nos han dado un p-value por debajo de 0,09, son los factores que consideramos que probablemente son factores de transcripción de la región promotora del gen AML1. Son AP-1 [T00029] y NF-kappaB [T00590].
Para este análisis usamos la misma secuencia promotora de AML1 en fasta.
| Nombre del factor | Posición inicial | Posición final | Secuencia | RE quality | RE query |
|---|---|---|---|---|---|
| IRF-1 [T00423] | 486 | 494 | TTTCCCTTT | 0,0042 | 0,00647 |
| IRF-1 [T00423] | 574 | 582 | TTTCCCTTT | 0,0042 | 0,00647 |
| NF-AT2 [T01945] | 481 | 490 | AATTCTTTCC | 0,00839 | 0,01178 |
| NF-AT1 [T00550] | 482 | 490 | ATTCTTTCC | 0,01259 | 0,01932 |
| HNF-3alpha [T02512] | 503 | 510 | TTAAAATA | 0,01678 | 0,0738 |
| Elk-1 [T00250] | 912 | 920 | GGAAGGAAG | 0,01678 | 0,01415 |
| SRY [T00997] | 292 | 300 | CTTTGTTTT | 0,03357 | 0,05427 |
| PEA3 [T00685] | 666 | 674 | CCACATCCT | 0,03777 | 0,03481 |
| LEF-1 [T02905] | 798 | 805 | CAGCAAAG | 0,05035 | 0,05021 |
| HNF-3alpha [T02512] | 738 | 745 | TCAAAATA | 0,05035 | 0,17214 |
| AP-1 [T00029] | 827 | 835 | ACTGAGTCA | 0,05035 | 0,05687 |
| PXR-1:RXR-alpha [T05671] | 190 | 197 | TGAACTAG | 0,06714 | 0,07102 |
| PR B [T00696] | 1009 | 1015 | AACAGTT | 0,06714 | 0,10733 |
| PR A [T01661] | 1009 | 1015 | AACAGTT | 0,06714 | 0,10733 |
| GR [T05076] | 757 | 763 | CAAAAAC | 0,06714 | 0,10918 |
| TCF-4E [T02878] | 799 | 805 | AGCAAAG | 0,06714 | 0,07138 |
| c-Jun [T00133] | 829 | 835 | TGAGTCA | 0,06714 | 0,07102 |
| NF-Y [T00150] | 979 | 986 | GAACCAAT | 0,06714 | 0,08249 |
| NF-1 [T00539] | 266 | 273 | TGTCCCAA | 0,06714 | 0,04703 |
En esta tabla presentamos únicamente aquellos fatores de transcripción que han dado un REequally por debajo de 0,09. Si comparamos las dos tablas (la obtenida con el programa vs. la obtenida con PROMO), se observa que los datos y los valores no coinciden, esto es debido a que usan diferentes matrices de pesos.
Para caracterizar la región promotora de este gen hemos aplicado los mismo protocolos que en el apartado anterior:
Hemos hecho un programa en perl*, para analizar la secuencia promotora de MTG8. Nos ha dado unos resultados que mostramos resumidos en la siguiente tabla:
*Visualizar programa en pantalla.
| Nombre matriz | Score | Posición | p-value |
|---|---|---|---|
| AP-1 [T00029] | -994.1866 | 425 | 0.45 |
| AR [T00040] | 3.0428 | 421 | 0.05 |
| c-Myc [T00140] | -1993.2437 | 463 | 0.95 |
| NF-AT1 [T00550] | -996.9756 | 86 | 0.72 |
| NF-kappaB [T00590] | -999.4796 | 62 | 0.38 |
| SRF [T00764] | -998.3631 | 187 | 0.32 |
| YY1 [T00915] | 3.3717 | 258 | 0.23 |
| RXR-alpha [T01345] | 1.8360 | 163 | 0.64 |
| HIF-1 [T01609] | -1995.4683 | 23 | 0.43 |
| AhR [T01795] | -997.3870 | 26 | 0.8 |
| PU.1 [T02068] | -996.2622 | 418 | 0.43 |
| HNF-4 [T02758] | -1995.4146 | 521 | 0.97 |
| NRSF [T06124] | -1993.9293 | 274 | 0.23 |
Marcamos en un color más claro aquellos factores que nos han dado un p-value por debajo de 0,09, que serán los factores que consideramos que probablemente son factores de transcripción de la región promotora del gen MTG8. Sólo hemos encontrado uno: AR [T00040].
Para este análisis usamos la misma secuencia promotora de MTG8 en fasta.
| Nombre del factor | Posición inicial | Posición final | Secuencia | RE quality | RE query |
|---|---|---|---|---|---|
| ELF-1 [T01113] | 67 | 79 | TTCTAGGAAGTAA | 0,00118 | 0,00067 |
| IRF-1 [T00423] | 976 | 984 | TTTCCCTTT | 0,0042 | 0,00701 |
| c-Fos [T00123] | 772 | 781 | GAGTCAGATG | 0,00629 | 0,00266 |
| PPAR-alpha:RXR-alpha [T05221] | 1033 | 1043 | CTGTCCCAGTC | 0,00787 | 0,00151 |
| HOXD9 [T01424] | 361 | 370 | AATTTATATT | 0,01259 | 0,22139 |
| HOXD10 [T01425] | 361 | 370 | AATTTATATT | 0,01259 | 0,22139 |
| NF-AT1 [T01948] | 784 | 793 | TGGAAAATTA | 0,02098 | 0,03803 |
| c-Ets-2 [T00113] | 116 | 124 | TTCCTTTTG | 0,02518 | 0,05919 |
| STAT1beta [T01573] | 205 | 214 | ATTTCCTAGT | 0,02832 | 0,02654 |
| SRY [T00997] | 565 | 573 | CTTTGTTTT | 0,03357 | 0,06306 |
| SRY [T00997] | 981 | 989 | CTTTGTTGT | 0,03357 | 0,06306 |
| LEF-1 [T02905] | 981 | 988 | CTTTGTTG | 0,05035 | 0,03516 |
| AP-1 [T00029] | 769 | 777 | TTTGAGTCA | 0,05035 | 0,04481 |
| VDR [T00885] | 1017 | 1025 | CTGGTGAAC | 0,05875 | 0,0189 |
| c-Ets-1 [T00112] | 206 | 212 | TTTCCTA | 0,06714 | 0,1314 |
| c-Jun [T00133] | 771 | 777 | TGAGTCA | 0,06714 | 0,05292 |
| NF-Y [T00150] | 721 | 728 | AAACCAAT | 0,06714 | 0,08377 |
| c-Ets-2 [T00113] | 373 | 381 | TAAAAGGAA | 0,09232 | 0,0775 |
| c-Ets-2 [T00113] | 1052 | 1060 | AGAGAGGAA | 0,09232 | 0,0775 |
En este caso, tampoco coinciden los resultados obtenidos mediante ambos métodos, como ya hemos señalado antes, será probablemente porque las dos aplicaciones aplican criterios diferentes.
Volver a Caracterización de la región promotora del gen.
Aunque este gen puede dar lugar a varios trascritos, parece ser que comparten las mismas funciones, que detallamos a continuación.
Tabla 1: Funciones del gen AML1.
| Identificador GO | Función |
|---|---|
| GO:0001501 | Desarrollo esquelético |
| GO:0003677 | Unión a DNA |
| GO:0003700 | Actividad de factor de transcripción |
| GO:0005515 | Unión a proteína |
| GO:0005524 | Unión a ATP |
| GO:0005634 | Núcleo |
| GO:0006350 | Transcrpción |
| GO:0006355 | Regulación de la transcripción, DNA-dependiente |
| GO:0007275 | Desarrollo |
| GO:0016563 | Actividad de activador transcripcional |
| GO:0030097 | Hematopoiesis |
| GO:0030182 | Diferenciación neuronal |
| GO:0030854 | Regulación positiva de la diferenciación del granulocito |
| GO:0031404 | Unión a ion clorito |
| GO:0045766 | Regulación positiva de la angiogénesis |
| GO:0045944 | Regulación positiva de la transcripción desde el promotor de la RNApolimerasaII |
| GO:0048266 | Respuesta conductual al pánico |
| GO:0048666 | Desarrollo neuronal |
Según la bibliografía, AML1 es un factor de transcripción heterodimérico, que se une al core de varios enhancers y promotores, por lo que está implicada en la regulación de otros genes. La unión de AML1 al DNA es específica de secuencia, lo que nos indica que se une exclusivamente a una serie de promotores para regular la acciones de genes determinados, algunos de ellos implicados en el proceso hematopoyético. La expresión de AML1 parece ser constitutiva en varios estadíos de la diferenciación hematopoyética, ya que fue detectado en todas las lineas celulares hematopoyéticas examinadas, especialmente en los linajes mieloides. Nos gustaría resaltar el papel de este gen en numerosas trasnlocaciones (entre ellas la que da lugar a nuestra proteína de fusión), que han sido asociadas a diferentes tipos de leucemia.
Figura 20: Estructura de la proteína generada por AML1.

En esta figura, se observan los dominios más importantes de AML1. Destaca el dominio runt, que ya ha sido mencionado anteriormente. Es un dominio muy conservado. También contiene un dominio de transactivación, una seńal de unión a la matriz nuclear (NMTS) y dos dominios inhibitorios. AML1 forma un heterodímero con CBFbeta, que facilita la unión a DNA de forma eficiente.
Resumiendo, las funciones principales de AML1 son:
En el caso de MTG8, los dos transcritos que genera la secuencia tienen funciones diferentes, por ello, las detallamos por separado:
Tabla 2: Funciones de MTG8a (ENST00000360348).
| Identificador GO | Función |
|---|---|
| GO:0003700 | Actividad de factor de transcripción |
| GO:0005634 | Núcleo |
| GO:0006355 | Regulación de la transcripción, DNA-dependiente |
| GO:0008270 | Unión a iones de zinc |
Tabla 3: Funciones de MTG8b (ENST00000265814).
| Identificador GO | Función |
|---|---|
| GO:0003700 | Actividad de factor de transcripción |
| GO:0005515 | Unión a proteína |
| GO:0005634 | Núcleo |
| GO:0006091 | Generación de metabolitos precursores y energía |
| GO:0006350 | Transcripción |
| GO:0006355 | Regulación de la transcripción, DNA-dependiente |
| GO:0008270 | Unión a iones de zinc |
| GO:0016564 | Represión de la actividad transcripcional |
| GO:0042803 | Actividad de homodimerización de proteína |
| GO:0045444 | Diferenciación de adipocitos |
| GO:0046872 | Unión a iones metálicos |
| GO:0051101 | Regulación de la unión a DNA |
Estos datos sugieren que MTG8 también da lugar a factores de transcripción, en concreto, genera un factor de transcripción putativo con dedos de zinc, que además puede actuar como oncoproteína. MTG8 pertenece a la familia ETO (que era otro de los nombres que recibe la misma proteína), todos los miembros de esta familia presentan regiones de homología con Nervy (NHR, Nervy homology regions), evolutivamente muy conservadas.MTG8 tiene 4 dominios NHR:
Figura 21: Estructura de la proteína generada por MTG8.
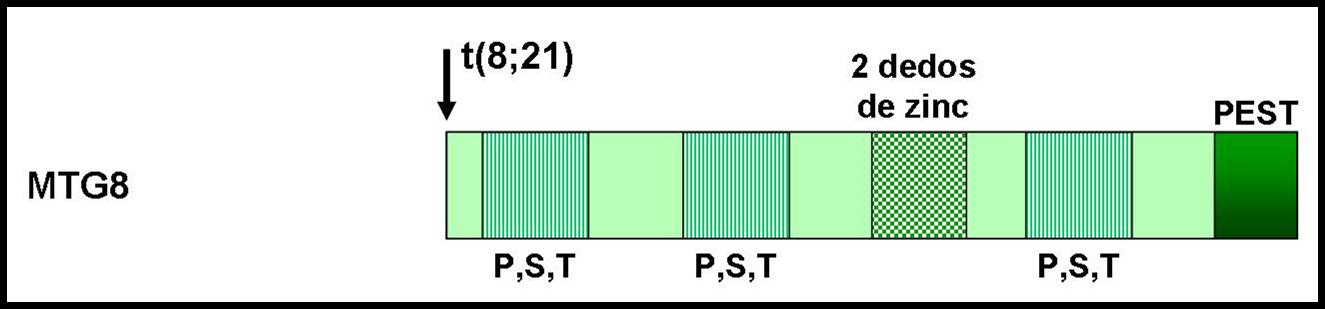
En la figura se observa un esquema de los principales dominios de MTG8, lo que da una idea de la función de dicha proteína. Como se señala en el párrafo anterior, se trata de un factor de transcripción. Tiene 3 regiones de secuencias ricas en prolina (P), Serina (S) y Treonina (T); estas regiones parecen estar implicadas en la regulación de MTG8, como posibles sitios de fosforilación, y también las zonas ricas en prolina en la activación de la transcripción. Por último, contiene una secuencia PEST en su extremo C-terminal, es una señal de degradación intracelular rápida de proteínas. Además hay evidencias que apuntan que MTG8 actúa como posible corepresor en la actividad represora de algunos factores de transcripción.
En cuanto a la proteína de fusión, conserva la mayor parte de la región N-terminal de AML1, con la región de homología con runt incluída, y la mayor parte de la proteína MTG8, como se observa en la siguiente figura.
Figura 22: Estructura de la proteína de fusión generada por la translocación t(8;21).
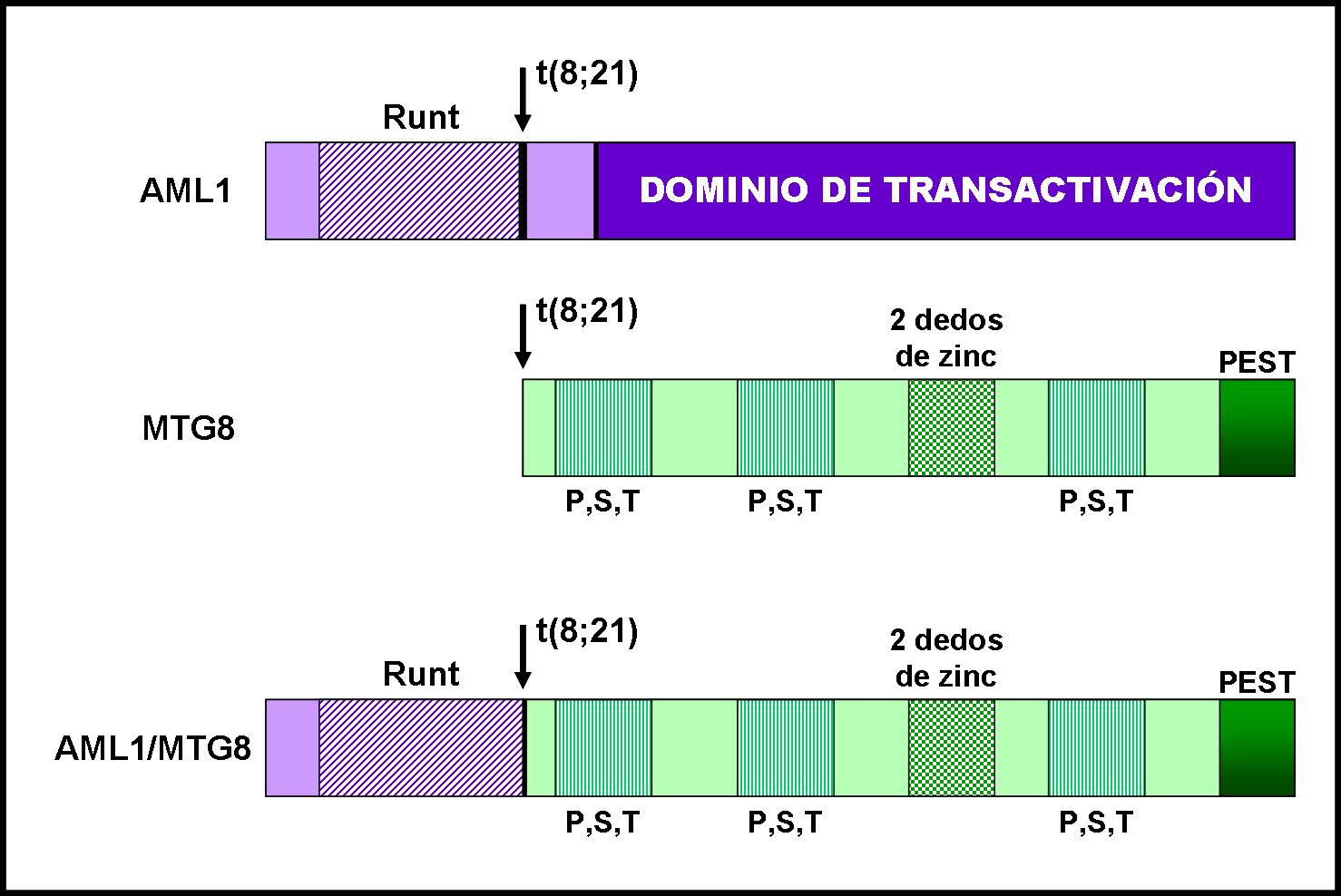
Como consecuencia de conservar el dominio runt, AML1-MTG8 se unirá a los promotores cuya actividad regula AML1, sobre los que acaba teniendo una actividad represora. Además, como AML1 y MTG8 no comparten las mismas localizaciones subnucleares, parece ser que el hecho de que AML1-MTG8 se colocalice con MTG8 hace que la relocalización de la actividad de unión a DNA de la porción AMT1 de la proteína de fusión actúe desregulando los genes diana de AML1, lo que podría explicar en parte su implicación en la patogésis de la leucemia mieloide aguda. Aún así, AML1-MTG8 también es capaz de transactivar otros genes que contienen sitios de unión a AML1. Por lo tanto, se ha observado que puede actuar como activador y como represor de la transcripción.
Por otra parte, parece ser que AML1-MTG8 es capaz de bloquear la diferenciación mieloide y eritropoyética. Esta proteína de fusión parece estar implicada en el corepresor nuclear o complejo de deacetilación de histonas, para bloquear la diferenciación hematopoyética.
A otro nivel, AML1-MTG8 ha mostrado un efecto negativo sobre la regulación del ciclo celular, incluyendo un arresto de ciclo en G1 asociado con niveles bajos de expresión de CDK4 y c-Myc.
Así, las diferentes observaciones descritas en la literatura sobre AML1-MTG8 demuestran que tiene efectos sobre los programas celulares relacionados con proliferanción, diferenciación y apoptosis. El potencial oncogénico de AML1-MTG8 reside en su capacidad para bloquear la diferenciación y promover la autorenovación de células madre y progenitores de la línea hematopoyética. En consecuencia, no cabe duda de la clara implicación de esta proteína en el proceso oncogénico que da lugar a la leucemia mieloide aguda.
De todo modos, cabe resaltar, que son necesarias mutaciones adicionales además de la translocación t(8;21), para que se produzca el desarrollo de la leucemia mieloide aguda.
Volver a Caracterización de la función del gen.
A lo largo de este estudio, hemos tratado de caracterizar la estructura genómica, la homología con otras especies, la expresión, la regulació y la función de los genes que dan lugar a la proteína de fusión AML1-MTG8. A partir de toda la información obtenida para los genes AML1 y MTG8, hemos podido deducir cómo es dicha proteína y a qué debe su implicación en el desarrollo de la leucemia mieloide aguda.
La proteína AML1-MTG8 se traduce a partir de un tránscrito de fusión producto de una translocación entre los cromosomas 8 y 21. Esta translocación, una de las aberraciones cromosómicas más frecuentes, hace que la región 5' del gen AML1 se fusione con la región 3' de MTG8, a la altura de un sitio de splicing alternativo. Dando lugar al DNA de fusión a partir del cual se expresará AML1-MTG8.
Ambos genes se encuentran muy conservados a lo largo de la evolución, lo que permite intuir la importancia de sus funciones en condiciones normales.
En cuanto a las diferencias de expresión, vemos que la distribución tisular de ambos genes es en cierto modo alterna, ya que AML1 se expresa mayoritariamente en timo, médula ósea y sangre periférica y no se expresa en el sistema nervioso central; mientras que MTG8 se expresa en Sistema Nervioso Central (Encéfalo y Médula espinal). Esto nos deja dudas en cuanto a dónde se expresa la proteína de fusión, pero lo que hemos encontrado en la literatura, parece indicar que principalente se conoce su expresión en células de las líneas hematopoyéticas, dada su implicación en la patogénesis d ela leucemia mieloide aguda.
En cuanto a la regulación de AML1-MTG8, sabemos que está controlada`por los promotores de AML1, ya que la parte procedente de MTG8 ha perdido su región 5'.
Por último, la función de la proteína quimérica deriva totalmente de la función que tení sus genes "progenitores", ya que tiene efectos sobre los programas celulares relacionados con proliferanción, diferenciación y apoptosis. Al conservar el dominio runt de unión a DNA de AML1, es capaz de interactuar con los genes que estaban bajo la regulación de AML1, tanto acitvándolos como reprimiéndolos. Estos genes están implicados en la regulación del proceso hematopoyético, este hecho unido a la posible expresión de AML1-MTG8 en células de la línea mieloide parece estar muy relacionado con su papel en el desarrollo de la leucemia mieloide aguda.
El potencial oncogénico de AML1-MTG8 reside pues en su capacidad para bloquear la diferenciación y promover la autorenovación de células madre y progenitores de la línea hematopoyética. Por lo que se deduce su importante implicación en el proceso neoplásico que da lugar a la leucemia. No obstante no se trata de un factor concluyente sino más bien capacitante, ya que es necesaria la adquisición de mutaciones adicionales además de la translocación t(8;21), para que llegue a producirse el proceso neoplásico que daría lugar al desarrollo de la leucemia mieloide aguda.
Por tanto, y a modo de conclusión podemos afirmar que AML1-MTG8 es una proteína oncogénica, implicada en el desarrollo de AML y producida por una aberración cromosómica: la translocación t(8;21)(q22;q22).
© Federica Eduati y Raquel Pinacho, 2007
{kind=link}
{kind=link}