We have found on the Ensembl Genome Browser Database that the DDIT3 gene only has a single transcript (Ensembl ID:ENST00000346473) which is translated into the 169 aminoacidic residues of the final protein, which sequence is represented below. As we can see on the transcript image, the DDIT3 gene is transcribed reversely (take a look at the sense of the transcription marked by the black arrow). Click on the Ensembl ID of DDIT3 to see the Ensembl Gene Report for ENSG00000175197.
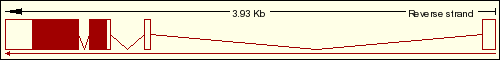
DDIT3 transcript
CHOP Protein Sequence:
MAAESLPFSFGTLSSWELEAWYEDLQEVLSSDENGGTYVSPPGNEEEESKIFTTLDPASLAWLTEEEPEP
AEVTSTSQSPHSPDSSQSSLAQEEEEEDQGRTRKRKQSGHSPARAGKQRMKEKEQENERKVAQLAEENE
RLKQEIERLTREVEATRRALIDRMVNLHQA
If we analyze the processed DDIT3 mRNA sequence which is showed below we will find that it has 4 exons in total, separated by black and blue colours. We also will realise that only the two last of them are codifying because the first translated codon ATG is on the start of the third exon and the TGA stop-codon is located in the middle of the fourth. We can see both codons marked in red bolds.
DDIT3 mRNA Sequence:
GAGGTCAGAGACTTAAGTCTAAGGCACTGAGCGTATCATGTTAAAGATGAGCGGGTGGCA
GCGACAGAGCCAAAATCAGAGCTGGAACCTGAGGAGAGAGTGTTCAAGAAGGAAGTGTAT
CTTCATACATCACCACACCTGAAAGCAGATGTGCTTTTCCAGACTGATCCAACTGCAGAG
ATGGCAGCTGAGTCATTGCCTTTCTCCTTCGGGACACTGTCCAGCTGGGAGCTGGAAGCC
TGGTATGAGGACCTGCAAGAGGTCCTGTCTTCAGATGAAAATGGGGGTACCTATGTTTCA
CCTCCTGGAAATGAAGAGGAAGAATCAAAAATCTTCACCACTCTTGACCCTGCTTCTCTG
GCTTGGCTGACTGAGGAGGAGCCAGAACCAGCAGAGGTCACAAGCACCTCCCAGAGCCCT
CACTCTCCAGATTCCAGTCAGAGCTCCCTGGCTCAGGAGGAAGAGGAGGAAGACCAAGGG
AGAACCAGGAAACGGAAACAGAGTGGTCATTCCCCAGCCCGGGCTGGAAAGCAGCGCATG
AAGGAGAAAGAACAGGAGAATGAAAGGAAAGTGGCACAGCTAGCTGAAGAGAATGAACGG
CTCAAGCAGGAAATCGAGCGCCTGACCAGGGAAGTAGAGGCGACTCGCCGAGCTCTGATT
GACCGAATGGTGAATCTGCACCAAGCATGAACAATTGGGAGCATCAGTCCCCCACTTGGG
CCACACTACCCACCTTTCCCAGAAGTGGCTACTGACTACCCTCTCACTAGTGCCAATGAT
GTGACCCTCAATCCCACATACGCAGGGGGAAGGCTTGGAGTAGACAAAAGGAAAGGTCTC
AGCTTGTATATAGAGATTGTACATTTATTTATTACTGTCCCTATCTATTAAAGTGACTTT
CTATGAGCC
In conclusion, we can affirmate that the total transcript has two non-coding exons (first and second) and two coding exons (third and fourth). And because of it only has one transcript on the Databases it only encodes for one isoform.
TLS/CHOP GENE
The TLS/CHOP gene is transcribed and translated to an hybrid protein that is made of the first part of FUS protein and the complete CHOP protein. We have analyzed more specifically it's mRNA in order to know which exons are conserved from the original genes and which aminoacids are derived from those exons.
First of all we are going to analyze the protein sequence of TLS/CHOP which is showed below. We have coloured the different regions that takes coincidences with the original proteins . We have marked in purple the aminoacids that TLS/CHOP shares with the FUS protein and in orange the aminoacids that TLS/CHOP shares with the CHOP protein. We can check that while the whole CHOP protein is conserved in the hybrid potein, only the first half of FUS is present on it, until the region encoded by the seventh exon.
TLS/CHOP Protein Sequence
MASNDYTQQATQSYGAYPTQPGQGYSQQSSQPYGQQSYSGYSQSTDTSGYGQSSYSSYGQSQNTGYGTQS
TPQGYGSTGGYGSSQSSQSSYGQQSSYPGYGQQPAPSSTSGSYGSSSQSSSYGQPQSGSYSQQPSYGGQQ
QSYGQQQSYNPPQGYGQQNQYNSSSGGGGGGGGGGNYGQDQSSMSSGGGSGGGYGNQDQSGGGGSGGYGQ
QDRGGRGRGGSGGGGGGGGGGYNRSSGGYEPRGRGGGRGGRGGMGGSDRGGFNKFGVFKKEVYLHTSPHL
KADVLFQTDPTAEMAAESLPFSFGTLSSWELEAWYEDLQEVLSSDENGGTYVSPPGNEEEESKIFTTLDP
ASLAWLTEEEPEPAEVTSTSQSPHSPDSSQSSLAQEEEEEDQGRTRKRKQSGHSPARAGKQRMKEKEQEN
ERKVAQLAEENERLKQEIERLTREVEATRRALIDRMVNLHQA
As we can see in the protein sequence represented above, there are several aminoacids in the middle of the sequence that seems that are not derived from any of the two original proteins. Because of that, we need to analyze the mRNA sequence of TLS/CHOP, extracted from the NCBI Database, which is showed below. There are two columns which shows the same sequence but each represents different things. On the one hand, the first column shows the total number of exons that the sequence has; we have marked them in blue and black colours to differenciate them. On the other hand, the second column shows the conservation of the sequence with the FUS mRNA sequence (marked in purple colour) and with the DDIT3 mRNA sequence (marked in orange colour). We also have marked in red bolds the starting-codon ATG and the stop-codon TGA used in the translation process in the sequence of both columns.
| TLS/CHOP mRNA Sequence (exons) |
TLS/CHOP mRNA Sequence (from FUS and DDIT3 alignment) |
ATGCTCAGTCCTCCAGGCGTCGGTGCTCAGCGGTGTTGGAACTTCGTTGCTTGCTTGC
CTGTGCGCGCGTGCGCGGACATGGCCTCAAACGATTATACCCAACAAGCAACCCAAAG
CTATGGGGCCTACCCCACCCAGCCCGGGCAGGGCTATTCCCAGCAGAGCAGTCAGCCC
TACGGACAGCAGAGTTACAGTGGTTATAGCCAGTCCACGGACACTTCAGGCTATGGCC
AGAGCAGCTATTCTTCTTATGGCCAGAGCCAGAACACAGGCTATGGAACTCAGTCAAC
TCCCCAGGGATATGGCTCGACTGGCGGCTATGGCAGTAGCCAGAGCTCCCAATCGTCT
TACGGGCAGCAGTCCTCCTACCCTGGCTATGGCCAGCAGCCAGCTCCCAGCAGCACCT
CGGGAAGTTACGGTAGCAGTTCTCAGAGCAGCAGCTATGGGCAGCCCCAGAGTGGGAG
CTACAGCCAGCAGCCTAGCTATGGTGGACAGCAGCAAAGCTATGGACAGCAGCAAAGC
TATAATCCCCCTCAGGGCTATGGACAGCAGAACCAGTACAACAGCAGCAGTGGTGGTG
GAGGTGGAGGTGGAGGTGGAGGTAACTATGGCCAAGATCAATCCTCCATGAGTAGTGG
TGGTGGCAGTGGTGGCGGTTATGGCAATCAAGACCAGAGTGGTGGAGGTGGCAGCGGT
GGCTATGGACAGCAGGACCGTGGAGGCCGCGGCAGGGGTGGCAGTGGTGGCGGCGGCG
GCGGCGGCGGTGGTGGTTACAACCGCAGCAGTGGTGGCTATGAACCCAGAGGTCGTGG
AGGTGGCCGTGGAGGCAGAGGTGGCATGGGCGGAAGTGACCGTGGTGGCTTCAATAAA
TTTGGTGTGTTCAAGAAGGAAGTGTATCTTCATACATCA
CCACACCTGAAAGCAGATGTGCTTTTCCAGACTGATCCAACTGCAGAGATGGCAGCTG
AGTCATTGCCTTTCTCCTTCGGGACACTGTCCAGCTGGGAGCTGGAAGCCTGGTATGA
GGACCTGCAAGAGGTCCTGTCTTCAGATGAAAATGGGGGTACCTATGTTTCACCTCCT
GGAAATGAAGAGGAAGAATCAAAAATCTTCACCACTCTTGACCCTGCTTCTCTGGCTT
GGCTGACTGAGGAGGAGCCAGAACCAGCAGAGGTCACAAGCACCTCCCAGAGCCCTCA
CTCTCCAGATTCCAGTCAGAGCTCCCTGGCTCAGGAGGAAGAGGAGGAAGACCAAGGG
AGAACCAGGAAACGGAAACAGAGTGGTCATTCCCCAGCCCGGGCTGGAAAGCAGCGCA
TGAAGGAGAAAGAACAGGAGAATGAAAGGAAAGTGGCACAGCTAGCTGAAGAGAATGA
ACGGCTCAAGCAGGAAATCGAGCGCCTGACCAGGGAAGTAGAGGCGACTCGCCGAGCT
CTGATTGACCGAATGGTGAATCTGCACCAAGCATGAACAATTGGGAGCATCAGTCCCC
CACTTGGGCCACACTACCCACCTTTCCCAGAAGTGGCTACTGACTACCCTCTCACTAG
TGCCAATGATGTGACCCTCAATCCCACATACGCAGGGGGAAGGCTTGGAGTAGACAAA
AGGAAAGGTCTCAGCTTGTATATAGAGATTGTACATTTATTTATTACTGTCCCTATCT
ATTAAAGTGACTTTCTATG |
ATGCTCAGTCCTCCAGGCGTCGGTGCTCAGCGGTGTTGGAACTTCGTTGCTTGCTTGC
CTGTGCGCGCGTGCGCGGACATGGCCTCAAACGATTATACCCAACAAGCAACCCA
AAGCTATGGGGCCTACCCCACCCAGCCCGGGCAGGGCTATTCCCAGCAGAGCAGTCAG
CCCTACGGACAGCAGAGTTACAGTGGTTATAGCCAGTCCACGGACACTTCAGGCTATG
GCCAGAGCAGCTATTCTTCTTATGGCCAGAGCCAGAACACAGGCTATGGAACTCAGTC
AACTCCCCAGGGATATGGCTCGACTGGCGGCTATGGCAGTAGCCAGAGCTCCCAATCG
TCTTACGGGCAGCAGTCCTCCTACCCTGGCTATGGCCAGCAGCCAGCTCCCAGCAGCA
CCTCGGGAAGTTACGGTAGCAGTTCTCAGAGCAGCAGCTATGGGCAGCCCCAGAGTGG
GAGCTACAGCCAGCAGCCTAGCTATGGTGGACAGCAGCAAAGCTATGGACAGCAGCAA
AGCTATAATCCCCCTCAGGGCTATGGACAGCAGAACCAGTACAACAGCAGCAGTGGTG
GTGGAGGTGGAGGTGGAGGTGGAGGTAACTATGGCCAAGATCAATCCTCCATGAGTAG
TGGTGGTGGCAGTGGTGGCGGTTATGGCAATCAAGACCAGAGTGGTGGAGGTGGCAGC
GGTGGCTATGGACAGCAGGACCGTGGAGGCCGCGGCAGGGGTGGCAGTGGTGGCGGCG
GCGGCGGCGGCGGTGGTGGTTACAACCGCAGCAGTGGTGGCTATGAACCCAGAGGTCG
TGGAGGTGGCCGTGGAGGCAGAGGTGGCATGGGCGGAAGTGACCGTGGTGGCTTCAAT
AAATTTGGTGTGTTCAAGAAGGAAGTGTATCTTCATACATCACCACACCTGAAAGCA
GATGTGCTTTTCCAGACTGATCCAACTGCAGAGATGGCAGCTGAGTCATTGCCTTTCT
CCTTCGGGACACTGTCCAGCTGGGAGCTGGAAGCCTGGTATGAGGACCTGCAAGAGGT
CCTGTCTTCAGATGAAAATGGGGGTACCTATGTTTCACCTCCTGGAAATGAAGAGGAA
GAATCAAAAATCTTCACCACTCTTGACCCTGCTTCTCTGGCTTGGCTGACTGAGGAGG
AGCCAGAACCAGCAGAGGTCACAAGCACCTCCCAGAGCCCTCACTCTCCAGATTCCAG
TCAGAGCTCCCTGGCTCAGGAGGAAGAGGAGGAAGACCAAGGGAGAACCAGGAAACGG
AAACAGAGTGGTCATTCCCCAGCCCGGGCTGGAAAGCAGCGCATGAAGGAGAAAGAAC
AGGAGAATGAAAGGAAAGTGGCACAGCTAGCTGAAGAGAATGAACGGCTCAAGCAGGA
AATCGAGCGCCTGACCAGGGAAGTAGAGGCGACTCGCCGAGCTCTGATTGACCGAATG
GTGAATCTGCACCAAGCATGAACAATTGGGAGCATCAGTCCCCCACTTGGGCCACA
CTACCCACCTTTCCCAGAAGTGGCTACTGACTACCCTCTCACTAGTGCCAATGATGTG
ACCCTCAATCCCACATACGCAGGGGGAAGGCTTGGAGTAGACAAAAGGAAAGGTCTCA
GCTTGTATATAGAGATTGTACATTTATTTATTACTGTCCCTATCTATTAAAGTGACTT
TCTATG |
When we analized the protein sequence (look before) we found that some residues weren't present on FUS protien, neither CHOP protein. But now when we compare the mRNA sequence of the hybrid gene with the mRNA of the original genes, the whole sequence alignes. It is due to the fact that these residues are encoded by the non-translated second exon of DDIT3 mRNA. Using the information provided by the right column of the table, we can see which region of TLS/CHOP mRNA sequence is derived from the FUS mRNA sequence and which from the DDIT3 mRNA sequence. Considering that the whole sequence contains 1682 nucleotides, the region compressed between the first nucleotide and the 877th is derived from the first part of the FUS mRNA sequence, whereas the other region (from 877th nucleotide to the last one) is derived from the DDIT3 mRNA sequence.
On the left column we can see that TLS/CHOP mRNA sequence contains 10 exons in total which all encode for the final protein, because the starting-codon ATG it's present on the first exon and the stop-codon TGA it's on the last one. If we relacionate the number of exons with the information provided on the right column, we will be able to realize that the final of the seventh exon of the TLS/CHOP mRNA sequence coincides exactly with the final conserved part of the FUS mRNA sequence, and also happens with the beginning of the eighth exon of TLS/CHOP mRNA sequence, which is the same as the beginning of the conserved part of the DDIT3 mRNA sequence. So now we can extract two conclusions about this coincidences:
- Exons of the TLS/CHOP gene compressed from the first to the seventh are derived from the exons of the FUS gene, whereas the other three located downstream are derived from the exons of the DDIT3 gene.
- The translocation point between the FUS gene and the DDIT3 gene to form the hybrid TLS/CHOP gene is located on the 877th position of the FUS mRNA sequence which finalizes with the seventh exon, and continues with the downstream DDIT3 mRNA sequence from the 102th position, which coincides with the beginning of the second exon of this mRNA sequence.
We can analyze in the next table the different exons of TLS/CHOP, from which original gene are derived, which are the positions that occupy in both genes and finally for which residues encode on the final protein.
| Exon number |
Exon sequence |
Exon position in TLS/CHOP gene |
Exon length |
Exon derivation |
Codifying aminoacid residues |
| 1 |
ATGCTCAGTCCTCCAGGCGTCGGTGCTCAGCGGTGTTGGAACTTC
GTTGCTTGCTTGCCTGTGCGCGCGTGCGCGGACATGGCCTCAAACG |
1-91 |
91 |
FUS gene Exon 1 |
MASND |
| 2 |
ATTATACCCAACAAGCAACCCAAAG |
92-116 |
25 |
FUS gene Exon 2 |
DYTQQATQS |
| 3 |
CTATGGGGCCTACCCCACCCAGCCCGGGCAGGGCTATTCCCAGCAG
AGCAGTCAGCCCTACGGACAGCAGAGTTACAGTGGTTATAGCCAGTC
CACGGACACTTCAGGCTATGGCCAGAGCAGCTATTCTTCTTATGGCC
AGAGCCAGAACA |
117-268 |
152 |
FUS gene Exon 3 |
SYGAYPTQPGQGYSQQSSQPYGQQSYSGY
SQSTDTSGYGQSSYSSYGQSQNT |
| 4 |
CAGGCTATGGAACTCAGTCAACTCCCCAGGGATATGGCTCGACTGGC
GGCTATGGCAGTAGCCAGAGCTCCCAATCGTCTTACGGGCAGCAGTC
CTCCTACCCTGGCTATGGCCAGCAGCCAGCTCCCAGCAGCACCTCGG
GAAG |
269-412 |
145 |
FUS gene Exon 4 |
TGYGTQS
TPQGYGSTGGYGSSQS
SQSSYGQQSSYPGYGQQPAPSSTSGS |
| 5 |
TTACGGTAGCAGTTCTCAGAGCAGCAGCTATGGGCAGCCCCAGAGTGG
GAGCTACAGCCAGCAGCCTAGCTATGGTGGACAGCAGCAAAGCTATGG
ACAGCAGCAAAGCTATAATCCCCCTCAGGGCTATGGACAGCAGAACCA
GTACAACAGCAGCAGTGGTGGTGGAGGTGGAGGTGGAGGTGGAG |
413-601 |
188 |
FUS gene Exon 5 |
SYGSSSQSSSYGQPQSGSYSQQPSYGGQQ
QSY
GQQQSYNPPQGYGQQNQYNSSSGGGGGGGGGG |
| 6 |
GTAACTATGGCCAAGATCAATCCTCCATGAGTAGTGGTGGTGGCAGTGG
TGGCGGTTATGGCAATCAAGACCAGAGTGGTGGAGGTGGCAGCGGTGGC
TATGGACAGCAGGACCGTGGAGGCCGCGGCAGGGGTGGCAGTGGTGGCG
GCGGCGGCGGCGGCGGTGGTGGTTACAACCGCAGCAGTGGTGGCTATGA
ACCCAGAGGTCGTGGAGGTGGCCGTGGAGGCAGAGGTGGCATGGG |
602-842 |
241 |
FUS gene Exon 6 |
GNYGQDQSSMSSGGGSGGGYGNQDQSGGGGSG
GYGQQDRGGRGRGGSGGGGGGGGGGYNRSSG
GYEPRGRGGGRGGRGGMG |
| 7 |
CGGAAGTGACCGTGGTGGCTTCAATAAATTTGGTG |
843-877 |
35 |
FUS gene Exon 7 |
GGSDRGGFNKFGV |
| 8 |
TGTTCAAGAAGGAAGTGTATCTTCATACATCACCACACCTGAAAGCAG |
878-925 |
48 |
DDIT3 gene Exon 2 |
VFKKEVYLHTSPHL
KAD |
| 9 |
ATGTGCTTTTCCAGACTGATCCAACTGCAGAGATGGCAGCTGAGTCATTG
CCTTTCTCCTTTGGGACACTGTCCAGCTGGGAGCTGGAAGCCTGGTATGA
GGACCTGCAAGAGGTCCTGTCTTCAGATGAAAATGGGGGTACCTATGTTT
CACCTCCTGGAAATGAAGAG |
926-1095 |
170 |
DDIT3 gene Exon 3 |
DVLFQTDPTAEMAAESLPFSFGTLSSWELEAWYED
LQEVLSSDENGGTYVSPPGNEE |
| 10 |
GAAGAATCAAAAATCTTCACCACTCTTGACCCTGCTTCTCTGGCTTGGCTG
ACTGAGGAGGAGCCAGAACCAGCAGAGGTCACAAGCACCTCCCAGAGCCC
TCACTCTCCAGATTCCAGTCAGAGCTCCCTGGCTCAGGAGGAAGAGGAGG
AAGACCAAGGGAGAACCAGGAAACGGAAACAGAGTGGTCATTCCCCAGCC
CGGGCTGGAAAGCAGCGCATGAAGGAGAAAGAACAGGAGAATGAAAGGAA
AGTGGCACAGCTAGCTGAAGAGAATGAACGGCTCAAGCAGGAAATCGAGC
GCCTGACCAGGGAAGTAGAGGCGACTCGCCGAGCTCTGATTGACCGAATG
GTGAATCTGCACCAAGCATGAACAATTGGGAGCATCAGTCCCCCACTTGG
GCCACACTACCCACCTTTCCCAGAAGTGGCTACTGACTACCCTCTCACTA
GTGCCAATGATGTGACCCTCAATCCCACATACGCAGGGGGAAGGCTTGGA
GTAGACAAAAGGAAAGGTCTCAGCTTGTATATAGAGATTGTACATTTATT
TATTACTGTCCCTATCTATTAAAGTGACTTTCTATG |
1096-1682 |
587 |
DDIT3 gene Exon 4 |
EESKIFTTLDPASLAWLTEEEPEPAEVTSTSQS
PHSPDSSQSSLAQEEEEEDQGRTRKRKQSG
HSPARAGKQRMKEKEQENERKVAQLAEENE
RLKQEIERLTREVEATRRALIDRMVNLHQA
|
In this table we have marked in blue bolds the starting-codon ATG and the stop-codon TGA in the first and last exon sequences, and also with red bolds the aminoacid residues which are shared between the different exons. The purple bolded ATG codon located on the nineth exon corresponds to the starting-codon of the DDIT3 gene.
There are some things we can see with the information provided by the table:
- The translocation point between the FUS exon 7 and the DDIT3 exon 2 derives to the formation of a new codon GTG (marked in orange bolds) which codifies for a valine residue. This valine is absent in the FUS protein and also in the translated-DDIT3 Exon 2 (which is normally untranslated).
- The DDIT3 exon 2 translocation into the codifying region of the FUS mRNA causes that this exon becomes codifying on the hybrid TLS/CHOP gene, which derives to a new aminoacid sequence located between the FUS protein region and the DDIT3 protein conserved in the hybrid protein.
We have made below those lines a similar version of the TLS/CHOP protein sequence we have presented before where we have marked in blue this new codified aminoacid sequence (by the original untranslated exons of the DDIT3 gene) and in red the valine which is codified by the new codon CTG generated by the translocation point:
TLS/CHOP Protein Sequence
MASNDYTQQATQSYGAYPTQPGQGYSQQSSQPYGQQSYSGYSQSTDTSGYGQSSYSSYGQSQNTGYGTQS
TPQGYGSTGGYGSSQSSQSSYGQQSSYPGYGQQPAPSSTSGSYGSSSQSSSYGQPQSGSYSQQPSYGGQQ
QSYGQQQSYNPPQGYGQQNQYNSSSGGGGGGGGGGNYGQDQSSMSSGGGSGGGYGNQDQSGGGGSGGYGQ
QDRGGRGRGGSGGGGGGGGGGYNRSSGGYEPRGRGGGRGGRGGMGGSDRGGFNKFGVFKKEVYLHTSPHL
KADVLFQTDPTAEMAAESLPFSFGTLSSWELEAWYEDLQEVLSSDENGGTYVSPPGNEEEESKIFTTLDP
ASLAWLTEEEPEPAEVTSTSQSPHSPDSSQSSLAQEEEEEDQGRTRKRKQSGHSPARAGKQRMKEKEQEN
ERKVAQLAEENERLKQEIERLTREVEATRRALIDRMVNLHQA
 Go back
Go back
CONSERVATION OF GENE AMONG SPECIES
In this section it's shown the information about the ortholog genes of FUS and DDIT3. Here are mentioned the species that have an ortholog gene of FUS or DDIT3, the Ensembl identification of the ortholog gene and the percentage of homology.
ORTHOLOG GENES OF FUS
| Picture |
Homo sapiens vs Specie |
Ensembl ID |
% homology peptide |
 |
Anopheles gambiae |
- |
43 |
 |
Caenorhabditis elegans |
- |
37 |
 |
Drosophila melanogaster |
- |
43 |
 |
Mus musculus |
- |
95 |
 |
Pan troglodytes |
- |
96 |
 |
Rattus norvegicus |
- |
94 |
 |
Saccharomyces cerevisiae |
- |
34 |
ORTHOLOG GENES OF DDIT3
| Picture |
Homo sapiens vs Specie |
Ensembl ID |
% homology peptide |
 |
Bos taurus |
ENSBTAG00000031544 |
92 |
 |
Canis familiaris |
ENSCAFG00000000232 |
92 |
 |
Cavia porcellus |
ENSCPOG00000012871 |
89 |
|
Caenorhabditis elegans |
- |
32 |
 |
Danio rerio |
ENSDARG00000059836 |
41 |
 |
Dasypus novemcinctus |
ENSDNOG00000017504 |
90 |
|
Drosophila melanogaster |
- |
30 |
 |
Echinops telfairi |
ENSETEG00000002521 |
57 |
 |
Erinaceus europaeus |
ENSEEUG00000003634 |
91 |
 |
Felis catus |
ENSFCAG00000002084 |
51 |
 |
Gasterosteus aculeatus |
ENSGACG00000006480 |
23 |
 |
Macaca mulatta |
ENSMMUG00000011286 |
98 |
|
Mus musculus |
ENSMUSG00000025408 |
88 |
 |
Oryzias latipes |
ENSORLG00000005361 |
24 |
|
Pan troglodytes |
ENSPTRG00000022781 |
99 |
|
Rattus norvegicus |
ENSRNOG00000006789 |
89 |
 |
Takifugu rubripes |
SINFRUG00000133607 |
24 |
 |
Tetraodon nigroviridis |
GSTENG00013575001 |
24 |
 |
Tupaia belangeri |
ENSTBEG00000000521 |
94 |
 |
Xenopus tropicalis |
ENSXETG00000006245 |
47 |
Go back
EXPRESSION CARACTERITZATION
As we have said before FUS is ligated downstream with DDIT3 to form the hybrid gene TLS-CHOP. Due to this we can confirm that the hybrid gene has the same promoter as FUS and the control of the expression of both genes will be similar. So to study the expression of TLS-CHOP we focus on the expression of FUS. Here are some pictures that show the expression of FUS in some experiments using different microarrays and tissues. If the gene has a high expression in the tissue it is shown in red, a basal expression is represented in black and a low expression in showed in green. We have made the same for the DDIT3 gene, in order to know how is it's basal expression and in which tissues it takes place.
FUS GENE
GNF Expression Atlas 2 Data from U133A and GNF1H Chips
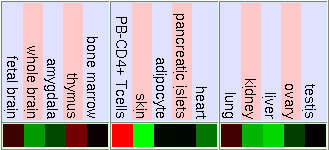
Normal Human Tissue cDNA Microarrays
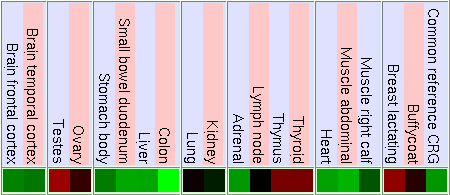
GNF Expression Atlas 1 Human Data on Affy U95 Chips
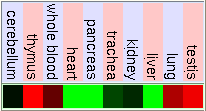
Gladstone Microarray Data Including Stem Cell Tissue

DDIT3 GENE
GNF Expression Atlas 2 Data from U133A and GNF1H Chips
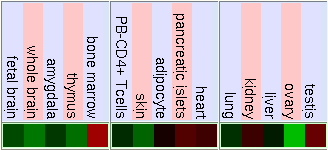
Normal Human Tissue cDNA Microarrays
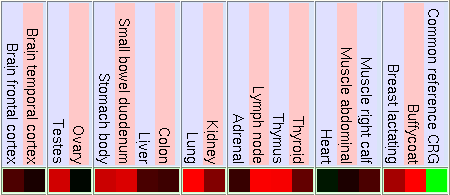
GNF Expression Atlas 1 Human Data on Affy U95 Chips
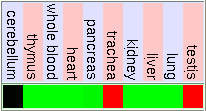
Gladstone Microarray Data Including Stem Cell Tissue

Go back
PROMOTOR REGION CARACTERITZATION
As we have said on the characterization of the genomic structure section, the hybrid gene TLS/CHOP contains at the beginning the genomic sequence of the FUS gene, so in order to study the promoter region of the TLS/CHOP gene we have to analyze which transcription factors are susceptible to bind to the promotor region of this gene.
The promoter sequence is represented below (which is extracted from the UCSC Genome Browser Database), which has marked in purple bolds the TSS (Transcription Starting Site) nucleotide, in blue the first exon and in red the first intron. The promoter region is written in small letters:
tttgcagttacaagacctggattcgaatcacgactcctcttagctgccctgtaatcaggcacaattacttgggtctctgagtctcactttccttatctag
aaaacggaggtatctttacttccttcgtaagactgatgacaaggaaattatctgtgcattttgaaaccacttaagccttgtacacgttttatttctggga
tcgccctggtagggcttcagaaaaataaaaaggaggtccctgagaaaaggctgggtaccgtacatctgaggtcaaccctctctggtcccaaggatggcct
gggctgttccgccccgtggctccccaggggcaaagccatgaggatccgggtgagagcccagtgctggacgagcccggggcccaggggtcccggccgaaat
ccctgctgtctttcaggtcaaacgtcataatccccgaaccccagaaaggccgaaaggcaaggcaaccctgaaagacgacgaagtcaacctcagggcgcag
gagagggagggccagtgtgctgccgacgagggaggctggagccgcggggacgaggcgccccatacagcggcaagagggtggagggcaggagctcgccatc
ctgggtgaaagcggggcccagcgaaggggcccggccacaggaatctcggttccaccccgctactcccggctgtgactccagtttcgtccccagccgccgg
gaccgccccctcgccccgcccccagcgggcactcaggccgtaccactgtgccttcatgggggtggagatagatcgtgggctagtcctgccgaggagagag
gggttcttcctcaaaaaatatgattatgtatagtattcgcatgattctagttaacttgtttcccttctgcctgctcggaccctctacctgccctacgaag
ggggcggagtgcgttcctgcctccccctgctcttccgcgtttggtgcgcgcctgcgcggtgcgtaggcggcggagcgtacttaagcttcgacgcaggagg
CGGGGCTGCTCAGTCCTCCAGGCGTCGGTACTCAGCGGTGTTGGAACTTCGTTG
CTTGCTTGCCTGTGCGCGCGTGCGCGGACATGGCCTCAAACGgtag
Once we had the promoter sequence isolated we had to introduce it on a Perl program made following the instructions on the web in order to know which transcription factors were able to bind into the sequence. This program is described on the Methods section. Below these lines we have presented the results of the program on a table format. We have for each transcription factor the region which it probably binds, it's punctuation and it's p value, which we have to consider as the probability to bind this region by a random situation. If a transcription factor has a low p value (it means that we have a low probability to bind by random that position with that TF) and a positive punctuation we can consider it as a good candidate to bind the promoter sequence of the FUS gene and obviously for the promoter sequence of TLS/CHOP gene.
| Transcription factor |
Start binding region |
End binding region |
Binding region sequence |
Punctuation |
p value |
| AP-1 [T00029] |
480 |
486 |
gaagtca |
2,651 |
0,24 |
| AR [T00040] |
1044 |
1050 |
GAACTTC |
2,747 |
0,43 |
| c-Myc [T00140] |
183 |
188 |
cacgtt |
-996,022 |
0,66 |
| NF-AT1 [T00550] |
143 |
149 |
ggaaatt |
3,201 |
0,21 |
| NF-kappaB [T00590] |
856 |
864 |
ttgtttccc |
-996.421 |
0,96 |
| SRF [T00764] |
751 |
759 |
ccttcatgg |
-996.355 |
0,43 |
| YY1 [T00915] |
294 |
299 |
atggcc |
2,678 |
0,58 |
| RXR-alpha [T01345] |
162 |
167 |
tgaaac |
2,496 |
0,64 |
| HIF-1 [T01609] |
1067 |
1075 |
TGCGCGCGT |
-996.58 |
0,99 |
| AhR [T01795] |
1072 |
1078 |
GCGTGCG |
3,011 |
0,29 |
| PU.1 [T02068] |
638 |
644 |
caggaat |
2,664 |
0,32 |
| HNF-4 [T02758] |
1042 |
1049 |
TGGAACTT |
-995.559 |
0,42 |
| NRSF [T06124] |
307 |
315 |
ttccgcccc |
-1994.202 |
0,91 |
As we have said before, the transcription factors that have positive punctuation and lower p value are good candidates to bind our sequence. So in this case we could consider the transcription factors AP-1, NF-AT1 or AhR as good options. However, we used another method to find these transcription factors, using the PROMO Database. Below these lines we have the results in a table format. Again, we are going to analyze the binding region of the same transcription factors used in our Perl Program and their p value, in this case represented by the RE query column. This term shows the probability to obtain the same results on another sequence with the same length and the same nucleotide proportions as ours.
| Transcription factor |
Start binding region |
End binding region |
Binding region sequence |
RE query |
| AP-1 [T00029] |
672 |
680 |
tgactccag |
0,07 |
| AR [T00040] |
1079 |
1087 |
GGACATGGC |
0,15 |
| NF-AT1 [T00550] |
142 |
150 |
ggaaattat |
0,03 |
| NF-kappaB [T00590] |
314 |
324 |
cgtggctcccc |
0,03 |
| YY1 [T00915] |
293 |
296 |
atgg |
4,011 |
| RXR-alpha [T01345] |
271 |
277 |
tcaaccc |
0,07 |
| AhR [T01795] |
1067 |
1076 |
GCGCGCGTGC |
0,03 |
First of all there are two things that we have to consider on the PROMO results:
- c-Myc, SRF, HIF-1, PU.1, HNF-4 and NRSF transcription factors are not found to bind the FUS promotor region because they don't appear on the PROMO results.
- There is a high numeric differency between the p values given by the Perl program and the RE query given by the PROMO results, but this is because both programs use a different type of matrix to analyze each transcription factor.
Now we have to compare both results to determinate which transcription factors are good candidates to bind the FUS promotor sequence. To do this comparation we have to ensure that the binding regions in the both results match approximately and the p value and the RE query should be similar. For example, the NF-AT1 transcription factor which has the lowest p value (0,21) also has a low RE query value (0,03), so it means that we have a low probability of random binding. In addition, the binding region of the promotor sequence is approximately the same in the Perl program and PROMO results. With all these data we can affirmate that the NF-AT1 transcription factor will probably bind the TLS/CHOP and FUS promotor sequences.
We also have another two transcription factors that could be considered good to bind the promotor sequence, such as AhR and YY1. The AhR transcription factor has low p and RE query values, and also would bind in a similar region in both results, the problem is that this binding region is located inside the first exon of the transcribed mRNA, so it really means that it's not a good option. On the other hand, the YY1 transcription factor matches the binding region in both results but has a very high RE query value, so it means that it has a high probability to bind the FUS or TLS/CHOP sequences by random.
To finalize this section we present again the TLS/CHOP (FUS) promotor sequence with the binding region of the NF-AT1 transcription factor marked in purple. We have also marked in orange the binding region of the other two transcription factors AhR and YY1:
tttgcagttacaagacctggattcgaatcacgactcctcttagctgccctgtaatcaggcacaattacttgggtctctgagtctcactttccttatctag
aaaacggaggtatctttacttccttcgtaagactgatgacaaggaaattatctgtgcattttgaaaccacttaagccttgtacacgttttatttctggga
tcgccctggtagggcttcagaaaaataaaaaggaggtccctgagaaaaggctgggtaccgtacatctgaggtcaaccctctctggtcccaaggatggcct
gggctgttccgccccgtggctccccaggggcaaagccatgaggatccgggtgagagcccagtgctggacgagcccggggcccaggggtcccggccgaaat
ccctgctgtctttcaggtcaaacgtcataatccccgaaccccagaaaggccgaaaggcaaggcaaccctgaaagacgacgaagtcaacctcagggcgcag
gagagggagggccagtgtgctgccgacgagggaggctggagccgcggggacgaggcgccccatacagcggcaagagggtggagggcaggagctcgccatc
ctgggtgaaagcggggcccagcgaaggggcccggccacaggaatctcggttccaccccgctactcccggctgtgactccagtttcgtccccagccgccgg
gaccgccccctcgccccgcccccagcgggcactcaggccgtaccactgtgccttcatgggggtggagatagatcgtgggctagtcctgccgaggagagag
gggttcttcctcaaaaaatatgattatgtatagtattcgcatgattctagttaacttgtttcccttctgcctgctcggaccctctacctgccctacgaag
ggggcggagtgcgttcctgcctccccctgctcttccgcgtttggtgcgcgcctgcgcggtgcgtaggcggcggagcgtacttaagcttcgacgcaggagg
CGGGGCTGCTCAGTCCTCCAGGCGTCGGTACTCAGCGGTGTTGGAACTTCGTTG
CTTGCTTGCCTGTGCGCGCGTGCGCGGACATGGCCTCAAACGgtag
Go back
As we said in the summary, we have studied the hybrid protein using the information provided by each original gene separately, because of the fact that we found less information of TLS/CHOP than those ones.
The function information of the three proteins (hybrid and the two originals) has been consulted in diverse literary sources that are mentioned in the References section.
All the information obtained to characterise the genomic structure of the hybrid gene involved in the myoxid liposarcoma (TLS-CHOP) was provided by UCSC genome browser, NCBI and Ensembl. We also used some blast tools to analyze the genomic comparations between the hybrid gene and the FUS and DDIT3 genes, such as Blastn and Blastp, and also the ClustalW software to make more specific study about the aminoacid conservation between the proteins. It is important to say that in order to make the ClustalW between the TLS/CHOP protein and the FUS protein we used the protein sequence of the first transcript of FUS, because it conserves the original exons and is not affected by the alternative splicing mechanism.
In order to study the gene conservation and know if FUS and CHOP are highly conserved among the species we have search for information in Ensembl, NCBI and UCSC. At the first genome browser mentioned, Ensembl, it is possible to find a lot of information of ortholog genes for DDIT3 but not for FUS. It's because of that we have looked for more information in other genome browsers. So the genes that don't have an ensembl ID are the homoleg genes that we have found in NCBI and UCSC.
To analyze the expression in diferent tissues of TLS-CHOP we have consulted the expression of FUS in UCSC. We also looked for the expression of DDIT3 just to compare it with the expression of FUS. In that genome browser it's possible to access to the Gene Sorter where some tables that show the expression are found. This expression were represented by a gradiation of colours. In order to make it more clear we gave an upper brigthness to the colours.
On the study of the promoter characterization section, we first had to found the 5' upstream region from the TSS (
Transcription Starting Site) of the FUS gene using the information provided by the USCS Genome Browser Database. We looked for the DNA sequence of the first 1000 nucleotides upstream region from the TSS of the FUS gene and the 100 nucleotides downstream region from it.
After that we programmed an algorism based in Perl language to be able to read a sequence (we use it with our promoter sequence) with the help of different TF matrix and find which of those transcription factors bind to the sequence. This program is based in four parts:
- The first part consists in reading the matrix for each TF and save them as a hash of vectors.
- The second part consists in transform this hash into a weight matrix, considering the nucleotide proportions in the sequence we have introduced.
- The third part consists in calculate the best punctuation of the matrix in every possible binding position of the transcription factor along the sequence.
- The final part consists in calculate the p value for each transcription factor, in where we consider the probability we have to find that TF in that position by random.
In addition, we have used the PROMO software. In the SelectSpecies section we only chase human factors and sites in order to reduce the number of TF obtained. However, we obtained so many TF so we decided to decrease the dissimilarity rate into 10 and give special attention only to those transcription factors that were analyzed with the Perl algorism.
Go back
To begin this section we are going to explain what we think it could happen with the hybrid protein TLS/CHOP. On the one hand, the FUS part doesn't conserve the RNA-binding region from the original protein so this is not able to stabilize the cellular mRNA when it's transcribed. This happens, as we said before, because this domain it's replaced by the CHOP DNA-binding and dimerization domains. As a result the FUS part of the hybrid protein would acquire a new function consisting in binding to another gene promoters using the CHOP DNA domain, causing the expression of oncogenic proteins which can help to develop the myoxid liposarcoma. On the other hand, in spite of the whole CHOP protein is conserved in TLS/CHOP, the fact that it is bound to the FUS protein and has translated it's second untranslated-exon would cause that the protein doesn't fold correctly and that would be the reason why it becomes to an afunctional transcription factor, incapable to bind it's normal target genes, which include the adipocyte growth and diferenttiation genes. This second fact also contributes to develope the disease.
We think that FUS has been discovered recently because as we said in the first section, it was named TLS when they found that it was involved in myoxid liposarcoma. Furthermore, there are other evidences like the fact that there are more ortholog genes for DDIT3 than for FUS. Actually in Ensembl database you will find lots of ortholog genes for CHOP but zero ortholog genes for FUS. We would affirmate that both genes are highly conservated in the mammal family (there is an exception on the conservation of the DDIT3 in the cat, only 51%), so it means that both play an important role in the organism. We can say that DDIT3 has an important function because not only controls the differentiation of an specific tissue, but also acts as a tumour supressor gene.
As we can see in the tables from expression caracteritzation, FUS is highly expressed in thymus, thyroid, CD4+ T cells and testis. It has a higher expression than the normal but not so intense in bood and lung. We can also apreciate that it has a low expression in lots of difernet organs like kidney, liver, heart, pancreas, skin, stomach and brain. The results of the expression of DDIT3 show that it also has a wide variety of expression on different tissues. DDIT3 is highly expressed in testis, thymus, lymph node,stomach, lung, trachea and bone marrow. It is shown that is has a low expression in ovary, heart, liver, kidney, blood and pancreas. We think that it could be related to the fact that these organs have a low composition of fat tissue. As CHOP is involved in cell division control and FUS stabilizes the mRNA it is natural that they have such an extensive expression.
We have used different strategies to find the TF that bind to the TLS-CHOP promoter. This results aren't quite reliable because the two methods used don't match exactly in all the FT. There is only one transcription factor that matches more or less in both methods and is suceptible to bind the promoter: NF-AT1. In addition, looking on the literature we found that there are some studies that demonstrated that there are three TFs that can bind the TLS-CHOP promoter. They are AP-2, GFC and Sp-1, so no one is NF-AT1. In the Perl program we have analyzed a variant of AP-2, the AP-1, which has a p value of 0,24, so we could consider this family of TF also good candidates.
Go back
- Crozac,A.,Aman,P.,Mandahi,N.,Ron,D. Fusion of CHOP to a novel RNA-binding protein in human myoxid liposarcoma. Nature363(6430):640-4.17 Jun 1993
- Thelin-Jarnum,S.,Lassen,C.,Panagopoulos,I.,Mandahl,N.,Aman,P. Identification of genes differentially expressed in TLS-CHOP carrying myoxid liposarcomas. Int J Cancer83(1):30-3.24 Sept 1999
- Rabbitts,TH.,Foster,A.,Larsson,R.,Nathan,P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet.4(2):175-80.Jun 1993
- Atlas of Genetics and Cytogenetics in Oncology and Hematology
- Online Book: Cancer Medicine. 6th ed. Kufe, Donald W.; Pollock, Raphael E.; Weichselbaum, Ralph R.; Bast, Robert C., Jr.; Gansler, Ted S.; Holland, James F.; Frei III, Emil, editors. Hamilton (Canada): BC Decker Inc; c2003
- OMIM data base: *137070 FUSION, DERIVES FROM 12-16 TRANSLOCATION, MALIGNANT LIPOSARCOMA;FUS
- Ensembl Genome Browser Database
- UCSC Genome Browser Database
- NCBI Database
- ClustalW
- PROMO Software
Go back