En nuestro proyecto hemos realizado una comparación de la estructura exónica de genes ortólogos de diferentes especies para estudiar el grado de conservación según la distancia evolutiva que las separe.
Para comenzar con una buena base...
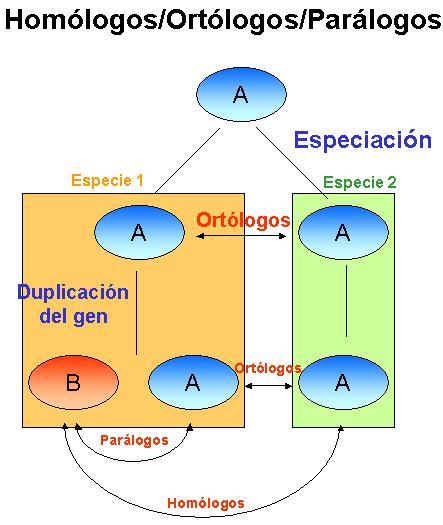
¿Que significa que dos genes sean ortólogos?
Para poder responder correctamente a esta pregunta deberemos hacer mención también de lo que significan los términos "homología" y "paralogía".A grandes rasgos, diremos que dos genes, o sus tránscritos, son "homólogos" cuando derivan de un ancestro común. Su similitud viene evaluada mediante los algoritmos de Smith-Waterman, BLAST o FASTA.
Estos genes serán "ortólogos" cuando esta similitud derive de la evolución de ellos en diferentes especies (relación vertical ). Mientras que serán considerados "parálogos" cuando esta similitud proceda de la duplicación del gen ancestral (relación horizontal ). En principio se asume que dos genes "ortólogos" tienen la misma función, mientras que los genes "parálogos", puesto que se han originado por duplicación y posterior divergencia, deben de tener diferente función o, al menos, cierto grado de especialización.
Esta clasificación no es totalmente estricta y, como casi todo, siempre hay lugar para algunas ambigüedades como el hecho de que existan genes ortólogos con funciones distintas y genes parálogos con una misma función.
Existen una serie de procesos evolutivos que pueden provocar que la transferencia de función entre proteínas ortólogas sea incorrecta y con ello que las funciones sean distintas a pesar de proceder de genes ortológos.
Existen otras clasificaciones, pero nosotras hemos utilizado la clásica para hacer referencia a los genes ortólogos.
Las especies con las que hemos trabajado en nuestro proyecto han sido:
Homo sapiens, Mus musculus, Gallus gallus y Tetraodon nigroviridis.
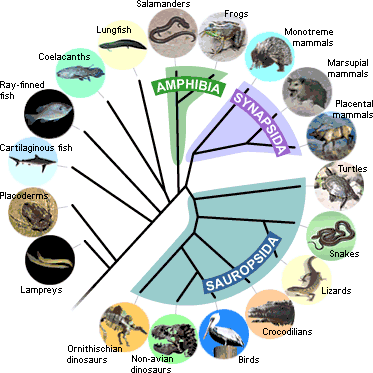
Para poder entender los resultados obtenidos de la comparación de la estructura exónica de los genes ortólogos comparados, deberemos tener presente cuál es la localización evolutiva de cada especie dentro del árbol evolutivo de los vertebrados, ya que cuanto más distancia evolutiva haya entre ellas, más probable será que la estructura exónica no esté tan conservada como la que esperaríamos entre especies cercanas.
Un árbol sencillo que nos puede ayudar a hacernos una idea aproximada podría ser el siguiente.
Observamos que de las tres especies que comparamos con humano, Tetraodon nigroviridis es la que se encentra a más distancia evolutiva, seguida por Gallus gallus y por último Mus musculus que está dentro del grupo de mamíferos, igual que Homo sapiens.
Por otro lado, las secuencias genómicas nos dan información de la estructura exónica. Al hablar de estructura exónica nos estamos refiriendo a la longitud de los exones que componen cada tránscrito y a las fases que definen los extremos 5' y 3' de cada uno de estos exones.
La conservación de la longitud de los exones de los tránscritos es importante, pero en muchos de los casos en que encontremos tránscritos ortólogos con uno o más exones de longitudes distintas, será a causa de la inserción o deleción de uno o más codones, con lo que la pauta de lectura no cambiará y el producto génico seguramente continurá siendo en esencia el mismo (a no ser que la mutación se de en un codón fundamental para la función de la proteína, pero nuestro proyecto se centra en la conservación de la estructura y no tiene en cuenta la secuencia de nucleótidos propiamente dicha).
Tal como hemos dicho, dentro de lo que llamamos "estructura exónica" también se incluyen los conceptos de las fases limitantes de los exones.
En principio, se espera que esta característica esté conservada, ya que un cambio de fase provoca un cambio del marco de lectura y una muy provable alteración del splicing alternativo de los intrones, y de la proteína resultante. En general, las mutaciones que provoquen cambios en el marco de lectura estarán selecionadas negativamente .
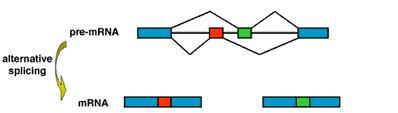
El splicing es el proceso necesario para la maduración del premRNA resultante directamente de la transcripción de la hebra del DNA. Tras el splicing, se obtiene un mRNA sin intrones que sólo conserva los exones codificantes que darán lugar a la proteína definitiva. El splicing, además de eliminar los intrones del tránscito, también puede eliminar algunos de los exones para dar lugar a toda una colección de tránscritos distintos que darán lugar a proteínas algo diferentes. Por eso se le llama "splicing alternativo", porque procesa diferentes tránscritos de forma alternativa.
Para que se dé el splicing existen unas determinadas secuencias que lo promueven, si el frame cambia, el splicing se verá alterado y por lo tanto, también afectará a la proteína final.
Así pues, para el proceso de splicing es importante conservar las fases de los extremos 5' y 3' de los exones delimitados por intrones.
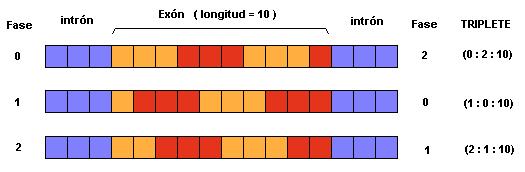
Las fases podrán ser 0, 1 o 2 dependiendo de la posición del intrón en relación con el marco de lectura del exón al que está delimitando.
El convenio de fases utilizado en este proyecto ha sido el siguiente:
Fase 0: Intrón localizado entre dos codones
Fase 1: Intrón interrumpiendo un codón entre la segunda y la tercera fase.
Fase 2: Intrón interrumpiendo un codón entre la primera y la segunda fase.