more
Treball Bioinformatica UPF. Multiple Sequence Alignment
Objective
The objective is to implement the progressive approach for multiple sequence
alignment in a simple PERL program.
Multiple Sequence Alignment
This is the approach implemented originally in the program ClustalW:
Thompson JD, Higgins DG, Gibson TJ (1994). ClustalW: improving the sensitivity
of progressive multiple sequence alignment through sequence weighting, position-specific
gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680.
You can find also information in http://genome.imim.es/courses/BioinformaticaUPF/T11/.
Introduction
The progressive global alignment approach works in the following way.
Given a set of N input sequences:
1. It builds a distance matrix from global pairwise alignments:
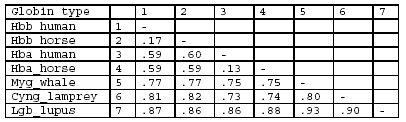
2. It builds a guide-tree from the mdistance matrix by the method of neighbour-joining (hierarchical clustering of sequences):
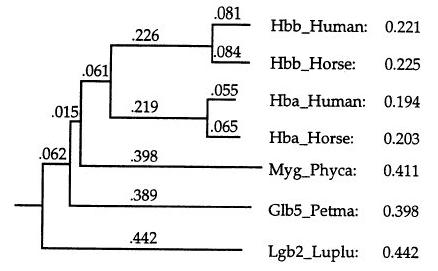
3. It performs a progressive alignment following the guide-tree; starting from the leaves sequences
and alignments are progressively aligned according to the structure of the tree:
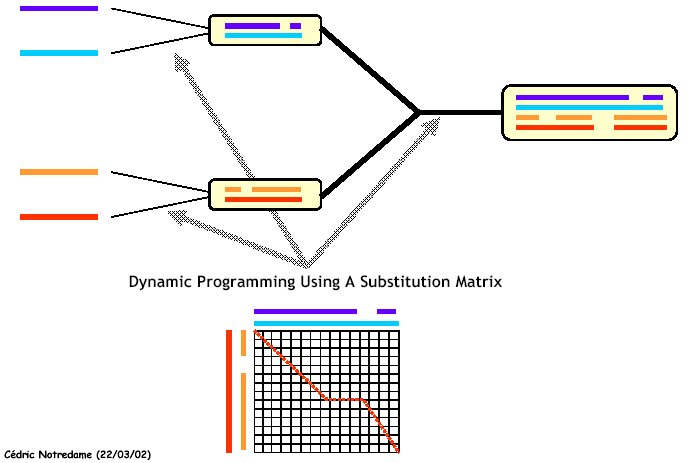
The main properties of this method are:
- Assigns weights to the sequences in order to correct the bias in the sampling of the evolutionary distances of the input sequences.
- It uses different substitution matrices in the different alignment steps according to the divergence rate of the
sequences to be aligned.
- Gap-penalty depends on the position: this allows gaps to form in less conserved regions (e.g. external loops of the protein structure), whereas
gaps are avoided in very conserved regions (e.g. core of the structure).
- Reconstructs dynamically the alingment adding new sequences.
Some potential pitfalls of this method are:
- Gaps are fixed after each alignment, hence there are strong dependencies on the choices in the initial alignments
and errors cannot be fixed at later stages.
Results can be viewed with other programs. For instance:
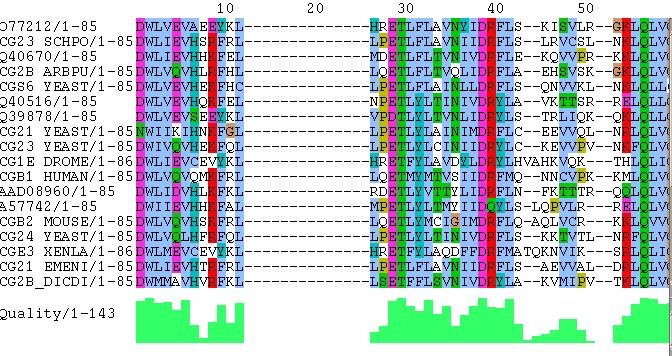
This multiple alignment snapshot from the alignment viewer Jalview highlights conserved
residues and conserved regions. More sophisticated patterns like
alternating hydrophobicity can be found in this way. This information
helps on the characterization of protein structure prediction. A
multiple alignment can also enable us to infer the evolutionary
history of the sequences. It is necessary to inspect long fragments in
order to make phylogenetic observations that are statistically
significant.
Design
This work is about developing a PERL program to perform a simple form
of multiple alignment. The method to be used must be the progressive
method described above. The design of the program should as modular
as possible so that extra functions can be easily added. For example:
gap penalties, different substitution matrices. The final
documentation must be written in HTML where the program and a
description of it are given.
The program should read a sequence file (multifasta) and the substitution matrix as input,
and give as output the multiple alignment.
Suggested work planning
- Write a PERL program to read two sequences (of proteins).
- Extend the program to perform the global pairwise alignment of two sequences using a substitution matrix (e.g. BLOSUM62).
- Extend the program to read more than two sequences.
- Introduce the first stage of the progressive alignment: create the distance matrix from all the pairwise alignments.
- Implement the alignment between 2 sequences (sequence-sequence), a sequence and a group of sequences (sequence-group) and between two groups of sequences
(group-group).
- Generate the progressive alignment (tree of alignments). The tree could be reported in plain text to the user as a series of alignments.
- Introduce an advanced gap and scoring schema.
- Create and return the output: a multiple sequence alignment of the input proteins. Use the ClustalW output as a guide. Colours are not necessary.
- Write some documentation about the program in HTML
Evaluation
A basic program implementing the progressive alignment and the
documentation are basic requirements. The additions like advanced gap
penalties, scoring or a web server schema will be positively
evaluated. The computational methods and the results should be
described in a simple web page or in an article format. It is
important that the work is presented in a way that is reproducible by
others.