|
INTRODUCCIÓN:
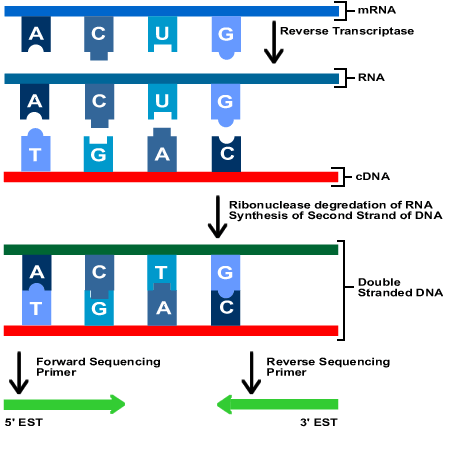
| Los EST o Expresed Sequence Tags, son secuencias simples de pequeños trozos de DNA, que son generados a partir de la secuenciación de uno o de los dos extremos del gen expresado. Su tamaño suele ser corto, de entre 200 y 500 nucleótidos y pueden ser secuencias codificantes o no codificantes. Para obtener EST partimos de células de las que extraemos el RNAmaduro y con primers y la transcriptsa reversa obtenemos su secuencia complementaria es decir el cDNA, que es mucho más estable que los mRNAs; se clonan en un vector, construímos una librería y finalmente los secuenciamos. De la secuenciación de la primera parte del cDNA obtenemos lo que conocemos como 5'EST y esta es la parte que pertenece a la región que suele codificar para proteinas y que, por lo tanto, es la parte que suele ser más conservada en una familia de genes. Si secuenciamos la parte final de la molécula de cDNA obtenemos los 3'EST que suelen ser partes no codificantes, o UTRs (untranslated regions), y que tienden a estar menos conservados. |
 |
La secuenciación de ESTs es rápida y barata y nos da información directa sobre la secuencia de los genes aunque esta información es parcial. Los resultados de trabajar con ESTs pueden ser muy diversos ya que podemos estar trabajando con un gen ya conocido, con uno similar a alguno conocido, puede ser un contaminante pero siempre cabe la posibilidad de que estemos delante de un nuevo gen. Pero la idea principal es secuenciar trozos de DNA que representen genes expresados en determinadas células, tejidos u órganos de diferentes organismos y utilizarlos para obtener información sobre un nuevo gen a partir de la similaridad de bases.
Actualmente se estan secuenciando nuevos genomas de diferentes especies pero en la mayoría de estos casos no tenemos ningún tipo de información sobre los genes que se estan expresando. Apis mellifera se encuentra en este caso y por esto se estan haciendo en paralelo estudios con EST de los cuales obtendremos toda la información sobre los genes expresados. Concretamente, se esta llevando a cabo un proyecto para acelerar el análisis molecular del comportamineto de las abejas en el que se trabaja con ESTs expresados en cerebro.
Las abejas són un importante modelo para el estudio de la plasticidad neural y del comportamiento, sobretodo lo relacionado con el comportamiento social, el aprendizaje y la memoria. Además las abejas son vitales en la comunidad agricultora como productoras de miel y como facilitadoras de la polinización. Finalmente, se utilizan como organismos modelos para estudiar inmunidad, reacciones alérgicas, resistencia a antibióticos y enfermedades del cromosoma X en humanos.
Para obtner más información del análisis de ESTs de abeja ver Este artículo
Proyecto de secuenciación de los ESTs de abeja Entra aquí
En esta web puedes encontrar información sobre la anatomía de la abeja melífera.
OBJETIVOS:
El objetivo principal del proyecto es analizar los ESTs de Apis mellifera , que hemos obtenido de una base de datos haciendo una comparación con genes humanos obtenidos de Ensemble.
A partir de esta comparación de secuencias, utilizando variables como el coverage y el porcentaje de identidad entre ambas secuencias, escogeremos el mejor match de cada EST. Mediante el identificador de la secuencia de gen humano podemos obtener información funcional y descripitva, entre otras, del gen en concreto. De esta forma podremos extrapolar esta información al EST que se ha alineado con el gen para ver si existe conservación funcional.
MATERIAL Y MÉTODOS:
El trabajo ha sido realizado con el sistema operativo UNIX, del cual hemos utilizado principalmente Emacs y Shell. Todos los programas han sido escritos con lenguaje PERL.
- Conseguir los ESTs de abeja que queremos estudiar y obtener los cDNA humanos de Ensembl, ambos en formato FASTA.
- Correr un TBLASTX, con un e-value de 0,01, para alinear los ESTs con los genes humanos, resultado que podemos ver en EST-human Blast. TBLASTX es una versión del BLAST en el que comparamos la secuencias, nucleótido a nucleótido.
- Calcular el coverage EST/cDNA así como el porcentaje de identidad. Para hacerlo realizamos diferentes pasos:
- Calcular longitud ESTs mediante el programa 1 obteniendo así un fichero con el identificador del EST y su correspondiente longitud.
- Mediante el programa 2 y el programa 3 conseguimos en un solo fichero el identificador del gen humano, el identificador del EST, el porcentaje de identidad y el coverage.
- A partir de este punto tenemos que decidir con que ESTs queremos seguir trabajando, para ello, seleccionamos, aquellos que tengan un coverage y un porcentaje de identidad mejores. Esto lo determinamos a partir de un umbral fijado en las gráficas de coverage y de identidad que son mayor del 80% y mayor del 60% respectivamente.
- Mediante el programa 4 , que considera los umbrales definidos anteriormente, logramos un listado que contiene solo los identificadores de EST y los de cDNAs con mejores resultados de coverage y un mayor porcentaje de identidad.
- A partir de esta lista, creamos un fichero que contiene sólo los identificadores de los cDNA y lo utilizamos para obtener información de estos genes utilizando Ensmart.
- Una vez obtenido este fichero lo juntamos, mediante el programa 5 , con el que contenía los dos identificadores; de este modo conseguimos el documento definitivo sobre el que trabajaremos. Este contiene: el identificador del gen, el identificador del EST, la función asociada y su localización anatómica.
- Con este fichero, buscamos la lista de los ESTs exclusivos de cerebro pero obtenemos una lista de ESTs demasiado grande y que además no son específicos de cerebro, es decir que se encuentran también en otras partes del organismo. Observando el fichero vemos que muchos de ellos son causantes de neoplasias y por ello decidimos quitarlos y trabajar sólo con aquellos ESTs del cerebro que no están asociados a neoplasias.
- Decidimos buscar ESTs específicos de una determinada zona del cerebro utilizando comandas como egrep y comm. El encéfalo es la única parte del cerebro donde hemos encontrado nueve ESTs que se expresan exclusivamente allí.
- A partir de estos ESTs y del archivo que contiene todos los genes del encéfalo conseguimos los correspondientes identificadores de genes humanos y su función.
- Utilizando el identificador de gen humano, buscamos información en diferentes bases de datos como Ensembl y Swissprot.
* Otros programas utilizados: TBLASTX, Parseblast.pl, R.
RESULTADOS:
Uno de los ficheros más importantes es el que obtenemos a partir del TBLASTX; este contiene el alineamiento de cada EST con una secuencia de cDNA humano. También nos da otros datos como el score, pero lo que nos interesa es el porcentaje de identidad que es uno de los valores utilizados para seleccionar el mejor alineamento.
Como hemos comentado anteriormente, para seleccionar el mejor alineamento nos hemos basado en unas gráficas realizadas a partir de los valores de coverage y del porcentaje de identidad que mostramos a continuación.
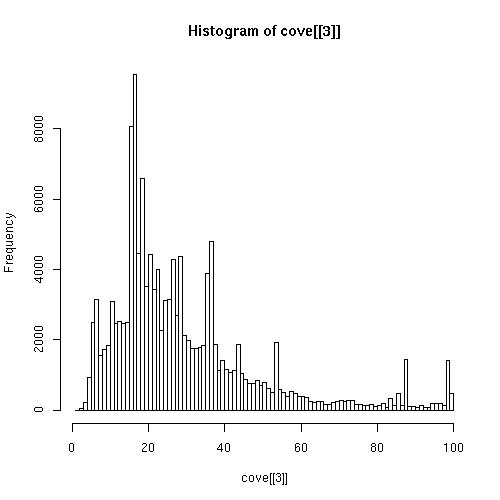 |
GRÁFICO 1
A partir del listado de EST-coverage y utilizando las comandas correspondientes del UNIX, conseguimos respresentar la frecuencia de EST en función del coverage que presentan. En esta gráfica vemos que la mayor parte de los EST se distribuyen alrededor de un valor random, que es del 20% de coverage, con ciertas variaciones. El pequeño grupo de secuencias que se halla a la derecha del valor random, es el que presenta un puntuación mayor, y por lo tanto son los que están más relacionadas con nuestras secuencias humanas. Por esto hemos definido el umbral a partir de un 80% de coverage, ya que creemos que estos serán los mejores EST para trabajar. |
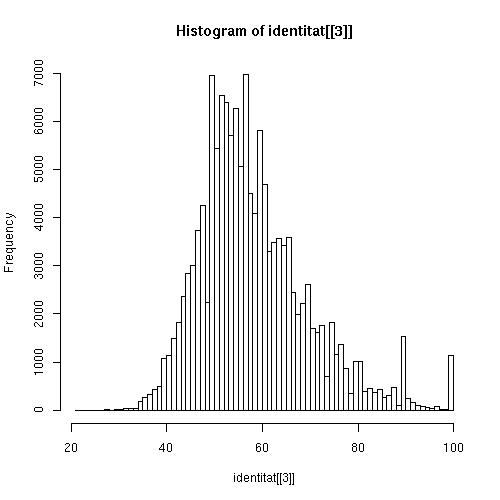 |
GRÁFICO 2
A partir del fichero que contiene EST-porcentaje de identidad y, utilizando también comandas del UNIX, representamos la frecuencia de EST que presentan un determinado porcentaje de identidad. En esta gráfica los mayores porcentajes de identidad se situan alrededor de 50. Por ello, hemos decidido establecer el umbral a partir del 60% de identidad. |
Otro fichero que queremos destacar es el que contiene los genes humanos con el EST que se ha alineado e información sobre la función y localización de este gen y, por lo tanto, que podría corresponder al EST.
En la siguiente tabla presentamos los 9 ESTs finales con su correspondiente identificador de gen humano y con su posible función asociada. Clicando sobre el gen podemos acceder al fichero del que proceden y clicando sobre Link obtenemos más información sobre el gen.
| BB160005A20G10 | ENSG00000013503.1 | RNA Polymerase III, Subunit 2 (RPC2) | |
| BB160013A10A03 | ENSG00000186907.1 | Reticulon 4 receptor-like 2 (RTN4RL2) | |
| BB160014B20B10 | ENSG00000129315.1 | Cyclin T1 | |
| BB160019A10F10 | ENSG00000170364.1 | Set domain and mariner transposase fusion gene | |
| BB160020B20F06 | ENSG00000065108.1 | Ganglioside induced differentiation associated protein 2 | |
| BB170008A10C08 | ENSG00000080802.4 | CCR4-NOT transcription complex, subunit 4 (CNOT4) | |
| BB170016B10H03 | ENSG00000021574.2 | Spastin
Fidgetin-like 1 (FIGNL1) |
|
| BB170029B10G04 | ENSG00000066926.1 | Ferrochelatase, mitochondrial precursor (Protoheme ferro-lyase)(Heme synthetase) | |
| BB170031A20F07 | ENSG00000115760.1 | Baculoviral IAP repeat-containing protein 6(Ubiquitin-conjugating BIR-domain enzyme apollon) |
CONCLUSIONES:
En este proyecto hemos trabajado con ESTs del cerebro de Apis mellifera, por lo tanto, en un principio esperábamos en los resultados, que la mayor parte de los ESTs se asociaran con genes humanos expresados en cerebro. Hemos comprobado que en realidad no es así. Nuestros ESTs se han alineado con cDNA que se halla en múltiples partes del organismo; obtenemos genes de expresión genital, endocrina, respiratoria, cardiovascular, urinaria, alimentaria, dermal, linforeticular o musculoesquelética. Probablemente esto sucede debido a la corta longitud de los ESTs utilizados. Al ser pequeños se alinean con cDNAs que no son su secuencia homóloga, sino que comparten alta similitud de bases.
El análisis del fichero que contiene únicamente los genes de cerebro, los ESTs y la anotación nos indica que no hay especificidad de lugar, es decir, que estos genes también se encuentran en otras zonas del organismo. Además el número de genes era demasiado elevado para hacer un estudio detallado.
Debido a esto hemos decidido profundizar en las anotaciones del gen y buscar genes específicos de áreas concretas del cerebro. Antes de hacer esto eliminamos los genes relacionados con neoplasias ya que nos podrían dar comparaciones funcionales erróneas.
Las zonas utilizadas para estudiar la expresión han sido cerebelo, líquido cefaloraquídeo, encéfalo, diencéfalo, médula oblongata, meninges, cerebro medio, sistema ventricular...Sólo en encéfalo hemos encontrado genes que se expresan exclusivamente allí y en ninguna otra parte cerebral.
De todos los ESTs analizados, son nueve los que se hallan en encéfalo. Estos presentan diferentes funciones, algunas de ellas todavía en investigación.
A continuación presentamos un listado del EST con su correspondiente identificador humano y la función que presenta este gen. De este modo se puede inferir la posible función de la secuencia que representa el EST.
- BB160005A20G10-ENSG0000013503.1: presentan un 94% de identidad y un 93,67% de coverage. Se trata de una RNApolimerasa dependiente de DNA que cataliza la transcripción de DNA a RNA usando los ribonucleotidos coma sustrato. En humano se localiza en el cromosoma 12.
- BB160013A10A03-ENSG00000186907.1: presentan un 80% de identidad y 81,03% de coverage. Es una secuencia homóloga al receptor NOGO-66 y se halla en el cromosoma 11.
- BB160014B20B10-ENSG00000129315.1: presentan un 76% de identidad y un 98,72% de coverage. Es la subunidad reguladora de la kinasa dependiente de ciclina CDK9 y por lo tanto es muy importante en ciclo celular. Se encuentra en el cromosoma 12 humano.
- BB160019A10f10-ENSG00000170364.1: tienen un 66% de identidad y un 89,58% de coverage. Se trata de un dominio SET y un gen de fusión a un transposón de tipo mariner. Se halla en el cromosoma 3.
- BB160020B20F06-ENSG00000065108.1: presentan un 72% de identidad y un coverage del 94,67%. Se trata de una proteina que induce diferenciación neuronal asociada a la proteina 2; presenta un ortólogo en ratón. Se encuentra en el cromosoma 1.
- BB170008A10C08-ENSG00000080802.4: presentan un porcentaje de identidad de 86% y 81,90% de coverage. Es la subunidad 4 de un complejo de transcripción que regula negativamente el proceso; esto se ha encontrado en levaduras. Se halla en el cromosoma 7.
- BB170016B10H03-ENSG00000021574.2: tienen un 80% de identidad y un 85,37% de coverage. Es probablemente un ATPasa involucrada en el ensamblje o función de complejos proteicos nucleares.Se halla en el cromosoma 2.
- BB170016B10H03-ENSG00000132436.1:presentan un porcentaje de identidad del 67% y 80,98% de coverage. Se trata de la proteina FIGNL1 codificada en el cromosoma 7.
- BB0023B10G04-ENSG00000066926.1: tienen un 72% de identidad y 86,71% de coverage. Se trata de una ferrocatalasa que cataliza la inserción de hierro en la porfirina 9. Se halla en el cromosoma 18.
- BB170031A20F07-ENSG00000115760.1: presentan un 87% de identidad y un 99,48%de coverage. Se trata de una proteina que protega a la célula frente la inducción de apoptosis. Se localiza en el cromosoma 2 humano.
REFERENCIAS:
http://www.ncbi.nlm.nih.gov/About/primer/est.htm
http://hgsc.bcm.tmc.edu/projects/honeybe
http://titan.biotec.uiuc.edu/bee/honeybee_project.ht
http://www.genome.org/cgi/content/abstract/12/4/55
http://www.molinicos.org/agricultura/La%20abeja%20IV.htm
http://www.ldc.usb.ve/~vtheok/webmaestro
http://html.conclase.net/tutorial/html
- Hu,P., Wu,S., Sun,Y., Yuan,C.-C., Kobayashi,R., Myers,M.P., Hernandez,N.,
Characterization of human RNA polymerase III identifies orthologues for Saccharomyces cerevisiae RNA polymerase III subunits.
(2002) Mol. Cell. Biol. 22:8044-8055
- Wei,P., Garber,M.E., Fang,S.-M., Fischer,W.H., Jones,K.A.,A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. (1998) Cell 92:451-462
- Bieniasz,P.D., Grdina,T.A., Bogerd,H.P., Cullen,B.R., Analysis of the effect of natural sequence variation in Tat and in cyclin T on the formation and RNA binding properties of Tat-cyclin T complexes.(1999) J. Virol. 73:5777-5786
-Liu,H. Nakagawa,T. Kanematsu,T. Uchida,T. Tsuji,S. Isolation of 10 differentially expressed cDNAs in differentiated Neuro2a cells induced through controlled expression of the GD3 synthase gene.J. Neurochem. 72:1781-1790 (1999)
CONTACTA CON NOSOTRAS:
Las autoras de este proyecto somos Montse Roura y Marta Valiente, estudiantes de cuarto curso de Biología de la Universidad Pompeu Fabra de Barcelona.
Si tienes cualquier duda o quieres algun tipo de información puedes contactar con nosotras, escribinedo un mail a cualquiera de estas dos direcciones:
Muchas gracias por tu visita!!