Las familias de genes se reconocen a menudo usando similitudes y así encontrar relaciones a nivel de secuencia nucleotídica o aminoacídica. Los datos de la secuencia genómica proporcionan la información arquitectónica del gen no usada por los métodos convencionales de comparación de secuencias. En particular, muchos genes eucariotas están divididos en exones, delimitados por los intrones.
Los intrones se pueden clasificar según su posición relativa a la pauta de lectura del gen:
| 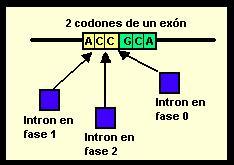 |
Se espera que las posiciones de los exones y fases de los intrones sean características relativamente conservadas entre genes homólogos, porque fusiones o divisiones de exones y cambios en la pauta de lectura se deberían seleccionar en contra.
Si consideramos un exon interno en una secuencia codificante, está limitado por un intron 5' y un intron 3'. El exon se puede describir por el trío:
Fase inicial del exón, Fase final del exón, Tamaño del exón.
Veámoslo con unos ejemplos:
Intron Exon Intron
...-------------...
5' Phase Phase Triplet
0 ...+++###+++###+... 1 (0,1,13)
1 ...++###+++###++... 2 (1,2,13)
2 ...+###+++###+++... 0 (2,0,13)
Pues en esto es en lo que se basa el Exon-Fingerprint.
Esencialmente un fingerprint es una manera diferente de representar un gen. El formato de un fingerprint debe ser completo pero sencillo, que permita un fácil análisis de los datos en él contenidos y que permita una comparación entre secuencias simple y eficaz.
La información que contiene un exon-fingerprint es:
Esta descripción simple de exon-fingerprint se complica para los exones iniciales y terminales, asíque vamos definir unas reglas especiales.
Dado que sólo consideramos los exones codificantes, es necesario tener en cuenta donde empieza y acaba el tránscrito. Por ejemplo:
start of translation (Met)
| end of translation (Stop)
____ ___|____ _________ _______|_______
transcript: |____|-------|___ATG__|------|_________|-------|_______TAG_____|
____ _________ ______
coding exons: |____|------|_________|-------|______|
Veamos un ejemplo de exon-fingerprint para el CDS del gen de la beta-globina de ratón:
J00413.1 3 3:2:92 2:0:223 0:3:129
J00413.1 es el identificador del gen para EMBL, tiene tres exones, el primero de los cuales, por ejemplo, tiene la fases 5' =3, la fase 3' = 2 y una longitud = 92bp. El fingerprint representa una secuencia codificante completa, y la fase 5' del primer exon y la 3' del último se les ha asignado el valor 3.
El objetivo de nuestro proyecto es desarrollar un programa que permita comparar los exon-fingerprint de tránscritos de dos especies estrechamente relacionadas. Inicialmente, necesitamos un programa que construya el exon-fingerprint de cada especie. [Esto corresponde al Programa1].
Después, usando las relaciones de ortología entre estos tránscritos, necesitaremos un algoritmo de comparación que correlacione cada par de ortólogos a sus respectivos exon-fingerprint. [Esto corresponde al Programa3].
Dado que estamos comparando tránscritos que han sido clasificados como ortólogos, esperamos que sus exon-fingerprints sean los mismos. Sin embargo hay ciertas variaciones, y es en función de estas variaciones que vamos a puntuar el aliniamiento.
Es mediante un Programa 4 que vamos a conseguir un aliniamiento global de los 'tripletes de símbolos'.
El sistema para puntuar la correspondencia exacta entre las fases de dos exones sería;:
Inicialmente teniendo en cuenta las diferencias en el tamaño del CDS y el número de exones, clasificamos los genes en cinco grupos:
Se considera que dos exones tienen tamaños distintos si difieren en más de 6 nucleótidos. Como consecuencia dos CDS tienen tamaños distintos, si la diferencia en el número de bases es superior al número medio de exones entre los dos genes multiplicado por 6.
El sistema de puntuar estos casos es multiplicar el valor del alineamiento según las fases por un 'bonus'.
Estos programas los hemos diseñado de manera que se puedan aplicar para cualquier combinación de dos especies.