Como todos sabemos, en el núcleo de la célula se encuetran todos los genes que codifican para la existencia del organismo, codifican las proteínas. Los genes se transcriben a mRNA (Transcription) y este se traduce a proteínas(Translation). La biología molecular se sustenta sobre esta base que aparentemente es bastante simple. Sin embargo no lo es lo es tanto.
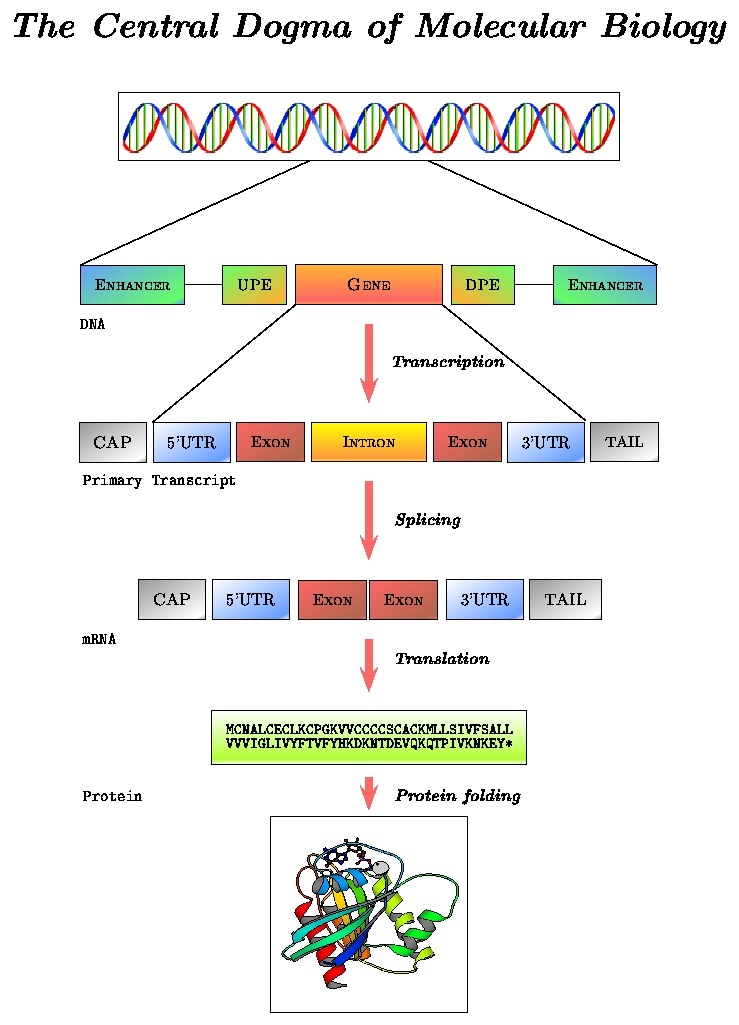
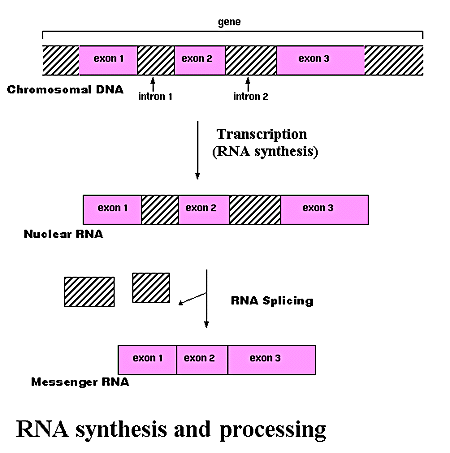
Los genes son entidades complejas, no todo el DNA de un gen es codificante, sólo una parte del DNA se transcribe para producir RNA nuclear, y sólo una pequeña parte de este sobrevive al procesamiento del RNA.
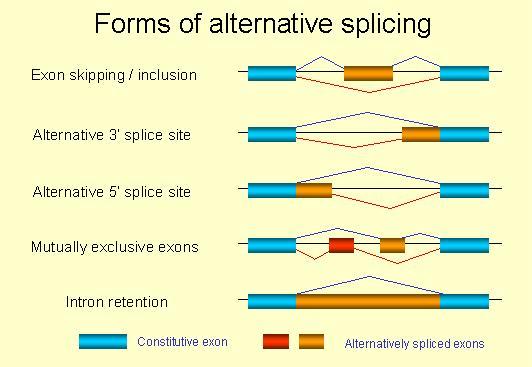
En muchos genes, hay regiones de la secuencia que no son codificantes (intrones), que se intercalan en regiones que sí lo son (exones). Uno de los pasos más importantes del procesamiento del RNA es el denominado splicing, que consiste precisamente en la eliminación de estos intrones formandose el denominado mRNA. Este proceso no tiene porqué conservar todos los exones, ni siquiera mantener el orden de estos, muchas veces se produce el llamado splicing alternativo, un proceso que permite que a partir de una misma secuencia de DNA puedan producirse un gran número de proteínas diferentes.
El mRNA sale del núcleo y viaja por el citoplasma donde se encontrará con los ribosomas, que son los encargados de su traducción a proteína.
La estructura exónica entre genes ortólogos de especies próximas suele estar bastante conservada, ya que modificaciones en la posición de los intrones o cambios de tamaño de los exones pueden tener consecuencias nefastas para la proteína. Los aminoácidos están codificados por tripletes de bases, codones, por lo tanto un cambio en la secuencia que implicase introducción o desaparición de nucleótidos en un número no múltiplo de 3 provocará un corrimiento del marco de lectura que afectará a todos los codones posteriores al lugar donde ha tenido lugar la modificación.
En general para estudios comparativos de genes, búsqueda de genes o proteínas homólogos o para estudios de filogénias, se suele utilizar la secuencia de nucleótidos o aminoácidos. Existen numerosos algoritmos informáticos de alineamiento que utilizan estas secuencias. El problema se da cuando se intenta, por ejemplo, alinear gran número de genes de tamaño considerable, ya que pese a haber sido optimizados al máximo, el proceso puede ser muy lento.
Ahora bien, se han propuesto otros sistemas de alineamiento que no impliquen necesariamente la secuencia, sino más bien la arquitectura del gen. Y es aquí donde entra en juego nuestro trabajo. Como ya hemos dicho la estructura exónica en principio debe estar bastante conservada entre genes ortólogos. Por tanto podríamos alinear de una manera bastante rápida y sencilla los exones en base a sus características, ya que el número de exones de un gen es abismalmente inferior a la cantidad de bases.
{kind=link}
{kind=link}