MATERIALSFormat de la taula d'ús de codons Format de les matrius de pesos
|
MÈTODESPautes obertes de lectura i pautes dels exons
|
DISSENY DEL NOSTRE PROGRAMA
|
El programa EXON-finding treballa amb dos formats de fitxers diferents, explicats a continuació.
Els fitxers en format FASTA són fitxers en què el seu nom té, o més aviat sol tenir, extensió .fa. Aquest format s'utilitza per enregistrar seqüències. Cada seqüència va precedida d'una línia que comença amb el símbol '>' i segueix una paraula que identificarà la seqüència. A partir de la línia següent trobem enregistrada la seqüència, típicament trencada en línies de 50, 60 o 70 símbols. Un exemple de fitxer en format FASTA podria ser el següent:
>NM_145815.chr19
ccaagtcctctgttctcaaactctgagcccaagggaaccccggccacatctcctccaaac
tgggggccccttcatttcccaggtctggatcgattcacttgccgggagagactttttaca
actcatctgcagctccgggtgcggttgggggagatagcgaagggtctggcctcgctgtga
tctgatttgggattaaaggtttggaaatttaa
>NM_145865.chr16
ctgcctggagagacatctggccaagttctggtgagcaggaaaaatgtctactcgttacca
cactcctctacattttgcagcctccaatggccatgcccactgcgtctcattcctggtcaa
ctttggtgccaacatctttgccctggataatgacttacagactccactggatgctgctgc
gggggccggctgctttctgctgctcacactgcct
Els fitxers en format GFF són fitxers el nom dels quals té extensió .gff. El propòsit d'aquest format és proporcionar una forma estàndard d'enregistrar les anotacions generades a partir de seqüències genòmiques. El seu contingut està organitzat en columnes de la següent manera:
on els valors no poden tenir cap espai tret de l'última columna, que a més a més és opcional, i estan separats entre si per una tabulació. La descripció de cadascun dels valors és la següent:
seqid source feature start end score strand frame [group]
...
seqidIdentificador de la seqüència a partir de la qual s'obté l'anotació (en el nostre cas, aquesta anotació correspon a la predicció d'un exó).
sourceOrigen de l'anotació, que en el nostre cas, correspon al nom que li hem donat al nostre programa (EXON-finding). En uns altres casos podria ser el nom de la base de dades de la qual prové l'anotació.
featureNom d'allò que estem anotant, o en unes altres paraules, tipus d'anotació. En el context d'aquest programa, hem utilitzat els següents termes: First (primer exó, extrem 5'), Terminal (últim exó, extrem 3'), i Internal (exó que es troba entre el primer i l'últim).
startPosició dins la seqüència on comença l'anotació. Serà sempre un nombre sencer entre 1 i la longitut de la seqüència i la posició referida formarà part de l'anotació. Aquesta posició ha de ser forçosament més petita o igual que a la posició especificada com a end. Això implica que les posicions de les anotacions fetes al strand negatiu han d'estar especificades respecte al strand positiu.
endPosició dins la seqüència on acaba l'anotació. Serà sempre un nombre sencer entre 1 i la longitut de la seqüència.
scorePuntuació associada a l'anotació. Pot ser qualsevol número real. És convenient utilitzar un nombre fix de xifres decimals (en el cas d'EXON-finding es dóna la puntuació amb 6 decimals). Si no hi hagués puntuació associada, hi escriurem el símbol del punt '.'.
strandSerà el símbol + si l'anotació ha estat feta llegint la seqüència de 5' a 3', o el símbol - si l'anotació ha estat feta llegint la seqüència de 3' a 5'. Si aquesta informació no és rellevant, escriurem el símbol del punt '.'.
framePauta de lectura en la qual s'ha fet l'anotació, serà 0, 1 o 2. Utilitzarem el símbol del punt '.' quan la pauta de lectura no sigui rellevant. La pauta 0 indicarà que la primera base de l'anotació correspon a la primera base d'un codó. La pauta 1 indicarà que la primera base de l'anotació és la segona base del seu codó. La pauta 2 indicarà que la primera base de l'anotació és la tercera base del seu codó.
[group]Aquest és un valor opcional, que pot estar format per lletres o números, i inclús espais, i que es sol fer servir per agrupar anotacions, com ara, exons que pertanyen a un mateix gen. A EXON-finding no s'inclou aquesta columna.
# # GFF_sample.gff # chr1 geneid Utr 150 500 1 + . "bio gene" chr1 geneid First 300 500 1 + 0 "bio gene" chr1 geneid Internal 750 1000 1 + 0 "bio gene" chr1 geneid Terminal 1250 1550 1 + 1 "bio gene" chr1 geneid Utr 1250 1800 1 + . "bio gene" # chr1 geneid Utr 2600 2880 1 - . "rev gene" chr1 geneid First 2600 2775 1 - 0 "rev gene" chr1 geneid Internal 2350 2500 1 - 1 "rev gene" chr1 geneid Terminal 2230 2250 1 - 0 "rev gene" chr1 geneid Utr 2000 2250 1 - . "rev gene"
Com es pot observar, també es poden incloure línies de comentari que començaran amb el símbol '#'.
Format de la taula d'ús de codons
Aquesta taula pot estar enregistrada en un fitxer de text com a parells de valors codó-proporció:
GGG 0.01708
GGA 0.01931
GGT 0.01366
GGC 0.02494
GAG 0.03882
GAA 0.02751
GAT 0.02145
GAC 0.02706
GTG 0.02860
GTA 0.00609
GTT 0.01030
GTC 0.01501
GCG 0.00727
GCA 0.01550
...
Format de les matrius de pesos
Les matrius de pesos estaran enregistrades de manera que cada fila especifiqui els pesos dels nucleòtids d'una posició determinada. El primer valor serà la posició, i els següents quatre valors seran els corresponents als nucleòtids, l'ordre dels quals vindrà especificat a la primera línia que començarà amb la paraula P0. L'última línia estarà formada per la paraula XX seguida de la posició on comença (o acaba) l'exó. Un exemple d'aquest format és la següent matriu de pesos per puntuar senyals de donors a seqüències d'ADN de l'organisme humà:
P0 A C G T
01 0.302 0.483 -0.305 -0.856
02 0.817 -0.667 -0.743 -0.474
03 -1.143 -0.782 1.123 -1.660
04 -9999 -9999 0.000 -9999
05 -9999 -9999 -9999 0.000
06 1.083 -2.097 0.135 -2.246
07 1.032 -1.093 -0.627 -1.111
08 -1.218 -1.479 1.257 -1.534
09 -0.411 -0.358 -0.136 0.492
XX 3
Senyals de splicing
Distingirem dos tipus de senyals de splicing: els donors i els acceptors. Els donors determinen l'extrem 5' dels introns, mentre que els acceptors determinen l'extrem 3' dels introns. Sota l'assumpció que tractem amb senyals de splicing canònics, l'extrem 5' d'un intró comença amb el dinucleòtid GT, i l'extrem 3' acaba amb el dinucleòtid AG. Aquests dos dinucleòtids no només es troben en els extrems dels introns, sinó que també es poden trobar en qualsevol punt al llarg de la seqüència. Això fa que els senyals de splicing, ja siguin donors o acceptors, estiguin formats per una cadena de nucleòtids, en la qual es troba el punt de tall GT o AG, i el contingut de la qual ajuda a la maquinària de splicing a determinar el dinucleòtid GT, o AG, en aquella posició concreta, com a punt de tall.
Per tal d'establir a on hi ha un senyal de splicing utilitzarem matrius de pesos (position weight matrices-PWMs): una pels senyals dels donors i una altra pels senyals dels acceptors. Aquestes matrius estaran enregistrades en fitxers de text d'un format determinat per tal que el nostre programa pugui llegir-les.
Considerem que tenim una matriu de pesos per identificar un senyal de splicing, donor o acceptor, que denotarem per M. Assumim que aquesta matriu està organitzada en 4 files, una per a cada nucleòtid A,C,G,T i m columnes, una per a cada posició del senyal, on les posicions aniran desde 0 fins a m-1. Ens referirem a una posició particular de la matriu indexant primer les files, i després les columnes. D'aquesta manera M(C-5) es refereix al pes associat al nucleòtid C quan el trobem a la sisena posició dins el senyal.
Considerem que tenim una seqüència emmagatzemada en un vector s, tal que si la seqüència té n bases, el vector estarà indexat per les posicions 0 a n-1, i ens referirem a una base en particular com, per exemple, s(7) que especifica la vuitena base de la seqüència emmagatzemada al vector s. D'aquesta manera, la puntuació d'un possible senyal de splicing serà calculada sumant els pesos de la matriu que corresponen a un tros de seqüència que comença a la posició p i que estem examinant com a possible senyal de splicing:
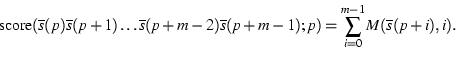
Donada una seqüència amb n bases podrem puntuar un màxim de n-m+1 possibles senyals de splicing. Decidirem quins d'aquests senyals poden ser realment considerats com a tals d'acord amb un llindar en la puntuació, tal que només aquells trossos de seqüència que donen una puntuació superior al llindar seran considerats com a senyals de splicing.
Els exons que es tradueixen a proteïna solen mostrar un biaix cap a l'utilització de determinats codons, depenent de l'organisme al qual la seqüència d'ADN pertany. Aquest fet permet una interpretació en termes estadístics de la composició de codons d'un exó codificant. Aquesta interpretació consisteix en assumir que la distribució dels codons observada als introns és uniforme, mentre que la distribució dels codons observada als exons codificants divergeix notablement d'aquesta uniformitat.
Aquesta divergència es determina mitjançant la ponderació dels codons observats a la seqüència que analitzem, d'acord amb una taula de l'ús dels codons a les regions codificants de l'organisme que estem analitzant. Donat un fragment de seqüència que es troba entre dos senyals de splicing, contra més codons observem que, d'acord amb la taula, apareixen freqüentment en regions codificants, direm que més gran serà el seu potencial codificant, i per tant serà més plausible que aquest fragment de seqüència correspongui a un exó.
Considerem una taula d'ús de codons que denotarem per T, i que estarà indexada pels codons de forma que T(ACT) es referirà a la proporció, en l'interval [0-1], d'ús del codó ACT en les regions codificants de l'organisme del qual s'ha derivat aquesta taula T. Assumim que tenim una seqüència emmagatzemada en format FASTA, i que volem calcular el biaix codificant d'una regió de q bases que va desde la posició p fins a la posició p+q-1 al vector s que emmagatzema la seqüència. Aquest biaix el calcularem com la raó de versemblança (o likelihood ratio) entre les proporcions esperades, que obtenim a partir de T, dels codons observats, i l'assumpció d'uniformitat per la qual la proporció esperada d'un codó qualsevol és 1/64:
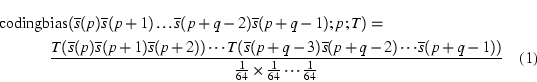
Els productes a l'expressió (1) donaran com a resultat números extremadament petits, per tant haurem de treballar amb el logaritme de l'expressió anterior.
Pautes obertes de lectura i pautes dels exons
Entenem per pauta de lectura, o reading frame, d'un exó la posició de la primera base de l'exó, dins el seu codó, considerant 0 com la primera posició. Si la primera base de l'exó, correspon a la primera base del codó al qual pertany, direm que la pauta és 0, si aquesta primera base del exó és la segona base del seu codó, direm que la pauta és 1, i finalment, si la primera base de l'exó és la tercera base del seu codó, direm que la pauta és 2.
Quan predim un exó cal anotar la seva pauta de lectura, a més a més de les corresponents posicions d'inici i final. Com que aquesta pauta ve determinada per la divisió dels nucleòtids en codons, caldrà primer determinar quines divisions en codons són correctes. Una divisió correcta en codons és aquella en què només podem trobar un codó de stop com a últim codú. Per tant caldrà trobar primer tots els possibles codons de stop i això ens determinarà el que es coneix com open reading frames (ORFs), o pautes obertes de lectura, que no són res més que parts de la seqüència que al dividir-les en codons, només podem trobar un codó de stop com a últim codú. Un cop tenim els ORFs, podrem anotar la pauta de lectura als exons que hem predit dins aquest ORF.
A més a més, respecte a l'exó d'inici, i a l'exó de final, del gen (coneguts com a First i Terminal, respectivament) cal tenir en compte que el primer estarà determinat per un senyal de donor aparellat amb un codó d'inici (ATG), mentre que el segon estarà determinat per un senyal de acceptor aparellat amb un codó de stop (TAA, TGA, TAG). Idealment, a part d'exons interns, hauríem d'intentar predir exons d'inici (First) i de final (Terminal).
Seqüència en format FASTA
Matriu de donors
Matriu d'acceptors
Taula d'ús de codons (coding bias)
Cut-off o valor llindar per les senyals
Nombre de prediccions per senyal
Cut-off o valor llindar de cada exó predit
Longitud mínima de la predicció
A continuació detallem les funcions utilitzades pel programa EXON-finding.
Per fer això hem de realitzar diferents passos:
Buscar pels diferentsframes (0, 1 o 2) tots els codons de stop que hi ha in frame.Els resultats dels diferents ORFs trobats s'emmagatzemen en un hash de hashs, %ORF, on les keys del hahs principal indexen el frame (0, 1 o 2) i dins de cadascuna d'aquestes trobem un altre hash, on cada key conté un array amb la posició d'inici i de final d'un ORF determinat.
Per cada frame, emparellar dos codons de stop consecutius que determinaran un ORF.
Els passos a realitzar són:
Determinar el tros de seqüència que conté un ORF determinat.En aquest punt hem de tenir en compte que si una senyal està massa aprop d'un codó de stop, no trobarem la senyal ja que no podrem aplicar la matriu de pesos. Per això, en funció de l'ORF que estem examinant (primer, últim o intern) i el tipus de senyal que estem buscant, modificarem la longitud de la seqüència a examinar per tal que es puguin aplicar les matrius.
Examinar aquesta seqüència i puntuar els diferents senyals mitjançant una matriu de pesos.
Ens quedarem amb aquells senyals que tinguin una puntuació superior al Cut-off (valor mínim o llindar) donat.
Els resultats obtinguts d'aplicar la funció s'emmagatzemen al hash %ORF, on per cada orf (indexat per la segona key)a més de l'inici i el final, guardarem també un hash amb els diferents donors trobats (a la posició 2 de l'array) i un hash amb els diferents acceptors (posició 3).
First: exó delimitat per un ATG i un donor siteAquesta funció l'executarem per cadascun dels tres frames i consta dels següents passos:
Internal: exó delimitat per un donor site i un acceptor site
Terminal: exó delimitat per un acceptor site i un codó de stop
Definim el tros de seqüència que hi ha en cada ORFPer cada senyal, predim tants exons com sigui possible fins a un màxim de prediccions determinat per l'usuari sempre i quan tinguin una longitud mínima també prèviament especificada.
Busquem els ATGs que hi ha in frame en cadascun d'ells
En el cas que hi hagi ATGs i donor sites en el mateix ORF definim exons First amb la funció par
En el cas que hi hagi acceptor sites i donor sites en el mateix ORF definim exons Internal amb la funció par
En el cas que hi hagi donor sites definim exons Terminal
Aquesta funció ens retorna tres arrays, on a cada posició trobem un array que conté la posició d'inici i de final d'exó, la puntuació d'aquest i el frame.
Aquesta funció s'executa per cadascun dels ORFs que tenim. Donades unes senyals d'inici, si n'hi ha de final d'exó i aquestes estan situades posteriors a les d'inici, emparellem una senyal d'inici amb diferents senyals de final fins a un màxim determinat per l'usuari.
Retorna un array amb els arrays dels diferents exons trobats a cada ORF que contenen l'inici, el final, la puntuació i el frame de l'exó predit.
Determinar el nombre de bases que hi ha entre l'inici de la seqüència i l'inici de l'ex&oacue;.
Mirar quin és el residu en dividir per tres el valor anterior, per tal de conèixer el frame de l'exó.