Predicció de gens ab initio
Per tal d'intentar predir els possibles gens en la nostra seqüència usarem programes de predicció de gens. És important destacar que en aquests programes hi introduïrem com a seqüència inicial l' output del RepeatMasker (repeat.seq1fm1.masked.txt)
Nosaltres em emprat quatre programes, als quals hem accedit a partir dels respectius servidors:
- GENSCAN (server) (seq1m.genscan)
- GENEID (server) (seq1m.geneid)
- FGENESH (server) (seq1softberry)
- GRAIL (server) (seq1grail)
Els quatre programes ens prediuen un nombre diferent de gens en la nostra seqüència . Així, el geneid prediu 7 gens, 5 en forward i dos en reverse (els gens 4 i 6) i 27 exons; el genscan, 9 ; el fgenesh, 6 gens (tres en forward i tres en reverse) i 29 exons, i finalment, el grail prediu 131 exons.
Per poder comparar millor les prediccions, seria molt útil poder visualitzar les diferents prediccions simultàniament en un mateix gràfic. Un dels programes de visualització és el gff2ps, que necessita que les dades li siguin introduïdes en format gff. El output del geneid ja està en aquest format. En els altres tres outputs, serà necessari aplicar uns scripts adequats a cada programa, per obtenir el gff.
Per exemple, en el cas del genscan, la conversió a gff es ralitza mitjançant les ordres :
- gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "hg_dna", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3, $7, $1;
}' seq1m.genscan | \
sed 's/\.[0-9][0-9]$//' > seq1m.genscan.gff
Un cop tenim els quatre documents en format gff,els visualitzem amb gff2ps.La comanda del shell és:
- $ export PATH=/disc8/soft/perl/bin/:/disc8/bin/:$PATH
- $ gff2ps seq1m.geneid.gff seq1m.genscan.gff seq1softberry.gff seq1grail.gff> .genepredictions.ps
- $ convert - rotate 90 ps a jpg
- $ kview jpg
Imatge dels gens predits pels quatre programes utilitzats:
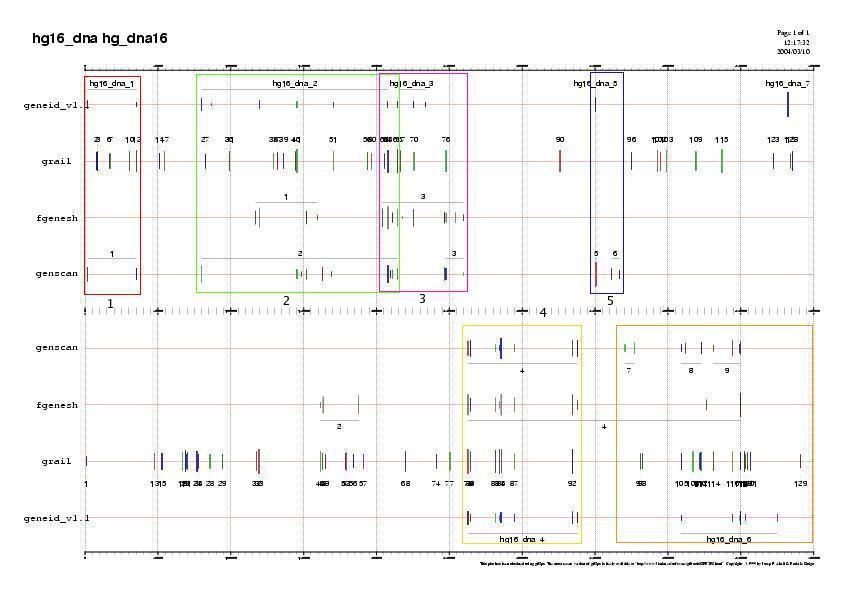
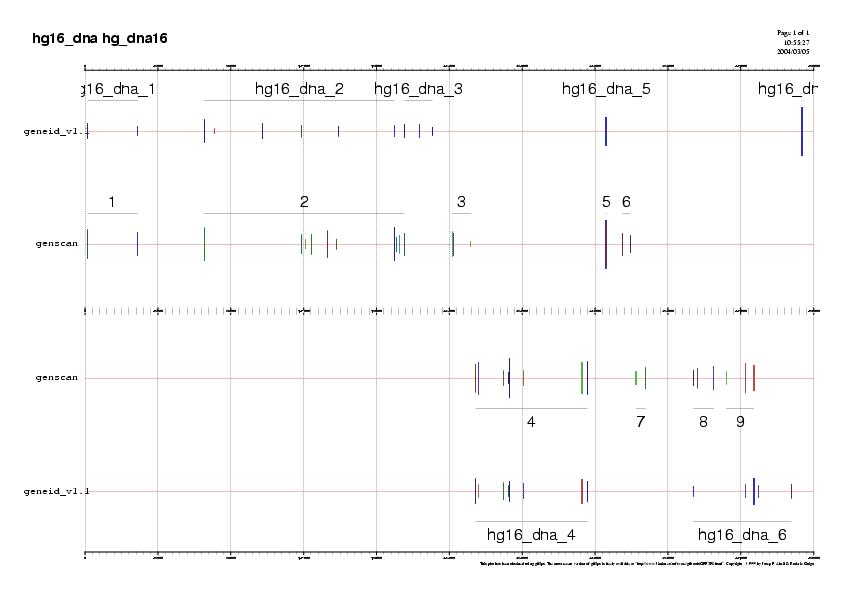
De l'observació del gràfic, deduïm que dues de les prediccions són més consistents entre elles: el geneid i el genscan. Fem un nou gràfic anàleg a l'anterior, però considerant només aquestes dues prediccions:
Un altre aspecte important a tenir en compte és si les repeticions que obtenim amb el RepeatMasker presenten una distribució homogènia al llarg de tota la seqüència. És a dir, si les repeticions es troben amb la mateixa freqüència en les regions exòniques i en les regions intròniques. En un principi, el que nosaltres esperem és que hi hagi més repeticions en les regions no codificants.
Per fer aquest gràfic necessitem disposar de la informació referent a les repeticions en una sola línia. Ho farem a partir d'un script usant gawk:
- gawk 'BEGIN{OFS="/t"}{$2="RepeatMasker"; print $0}' repeat.seq1fm.out.gff > repeat.seq1fm.out_singleline.gff
I visualitzarem els resultats amb kview:
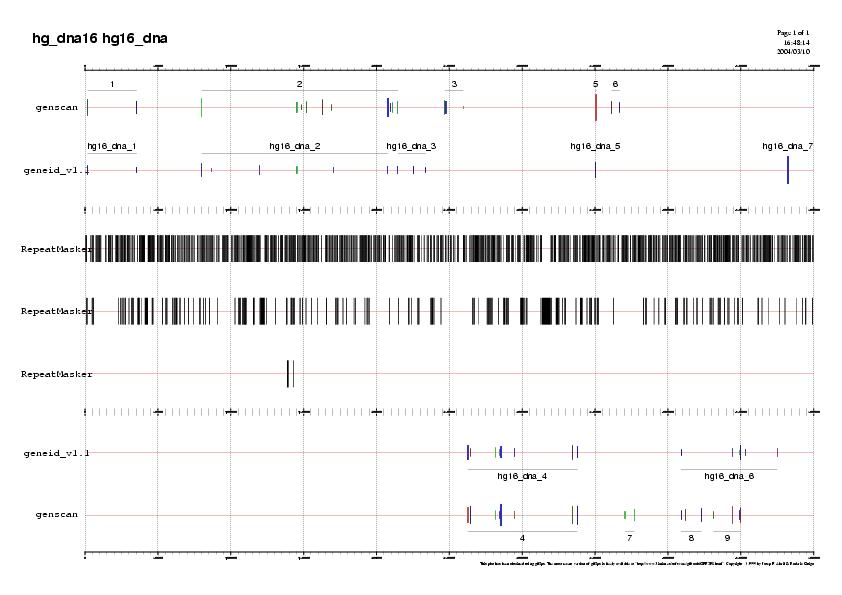
Distribució de les repeticions en les regions codificants i les no-codificants en geneid i genscan:
Distribució de les repeticions en les regions codificants i les no-codificants en tots els programes de predicció:
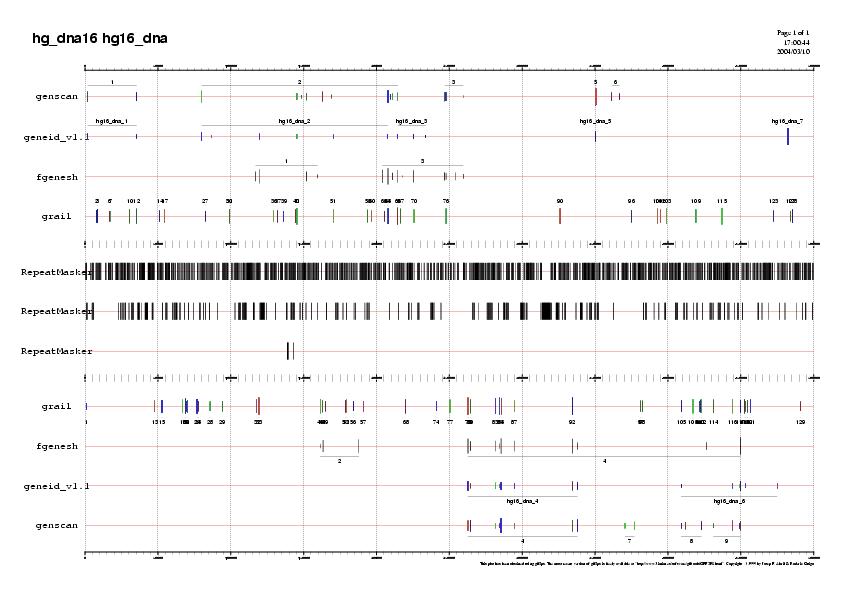
Tanmateix la densitat de les repeticions és tan elevada que no ens permet veure els resultats esperats.

Nosaltres em emprat quatre programes, als quals hem accedit a partir dels respectius servidors:
- GENSCAN (server) (seq1m.genscan)
- GENEID (server) (seq1m.geneid)
- FGENESH (server) (seq1softberry)
- GRAIL (server) (seq1grail)
Un cop tenim els quatre documents en format gff,els visualitzem amb gff2ps.La comanda del shell és:
- $ export PATH=/disc8/soft/perl/bin/:/disc8/bin/:$PATH
- $ gff2ps seq1m.geneid.gff seq1m.genscan.gff seq1softberry.gff seq1grail.gff> .genepredictions.ps
- $ convert - rotate 90 ps a jpg
- $ kview jpg
Imatge dels gens predits pels quatre programes utilitzats:
De l'observació del gràfic, deduïm que dues de les prediccions són més consistents entre elles: el geneid i el genscan. Fem un nou gràfic anàleg a l'anterior, però considerant només aquestes dues prediccions:
Un altre aspecte important a tenir en compte és si les repeticions que obtenim amb el RepeatMasker presenten una distribució homogènia al llarg de tota la seqüència. És a dir, si les repeticions es troben amb la mateixa freqüència en les regions exòniques i en les regions intròniques. En un principi, el que nosaltres esperem és que hi hagi més repeticions en les regions no codificants.
Per fer aquest gràfic necessitem disposar de la informació referent a les repeticions en una sola línia. Ho farem a partir d'un script usant gawk:
- gawk 'BEGIN{OFS="/t"}{$2="RepeatMasker"; print $0}' repeat.seq1fm.out.gff > repeat.seq1fm.out_singleline.gff
I visualitzarem els resultats amb kview:
Distribució de les repeticions en les regions codificants i les no-codificants en geneid i genscan:
Distribució de les repeticions en les regions codificants i les no-codificants en tots els programes de predicció:
Tanmateix la densitat de les repeticions és tan elevada que no ens permet veure els resultats esperats.
De l'observació del gràfic, deduïm que dues de les prediccions són més consistents entre elles: el geneid i el genscan. Fem un nou gràfic anàleg a l'anterior, però considerant només aquestes dues prediccions:
Un altre aspecte important a tenir en compte és si les repeticions que obtenim amb el RepeatMasker presenten una distribució homogènia al llarg de tota la seqüència. És a dir, si les repeticions es troben amb la mateixa freqüència en les regions exòniques i en les regions intròniques. En un principi, el que nosaltres esperem és que hi hagi més repeticions en les regions no codificants.
Per fer aquest gràfic necessitem disposar de la informació referent a les repeticions en una sola línia. Ho farem a partir d'un script usant gawk:
- gawk 'BEGIN{OFS="/t"}{$2="RepeatMasker"; print $0}' repeat.seq1fm.out.gff > repeat.seq1fm.out_singleline.gff
I visualitzarem els resultats amb kview:
Distribució de les repeticions en les regions codificants i les no-codificants en geneid i genscan:
Distribució de les repeticions en les regions codificants i les no-codificants en tots els programes de predicció:
Tanmateix la densitat de les repeticions és tan elevada que no ens permet veure els resultats esperats.
Un altre aspecte important a tenir en compte és si les repeticions que obtenim amb el RepeatMasker presenten una distribució homogènia al llarg de tota la seqüència. És a dir, si les repeticions es troben amb la mateixa freqüència en les regions exòniques i en les regions intròniques. En un principi, el que nosaltres esperem és que hi hagi més repeticions en les regions no codificants.
Per fer aquest gràfic necessitem disposar de la informació referent a les repeticions en una sola línia. Ho farem a partir d'un script usant gawk:
- gawk 'BEGIN{OFS="/t"}{$2="RepeatMasker"; print $0}' repeat.seq1fm.out.gff > repeat.seq1fm.out_singleline.gff
I visualitzarem els resultats amb kview:
Distribució de les repeticions en les regions codificants i les no-codificants en geneid i genscan:
Distribució de les repeticions en les regions codificants i les no-codificants en tots els programes de predicció:
Tanmateix la densitat de les repeticions és tan elevada que no ens permet veure els resultats esperats.
I visualitzarem els resultats amb kview:
Distribució de les repeticions en les regions codificants i les no-codificants en geneid i genscan:
Distribució de les repeticions en les regions codificants i les no-codificants en tots els programes de predicció:
Tanmateix la densitat de les repeticions és tan elevada que no ens permet veure els resultats esperats.