|
|
NCBI Conserved Domain Search |
| New Search | PubMed | Nucleotide | Protein | Structure | CDD | Taxonomy | Help? |
|
||||||||
|
 �� .. This CD alignment includes 3D structure. To display structure, download
Cn3D! �� .. This CD alignment includes 3D structure. To display structure, download
Cn3D!
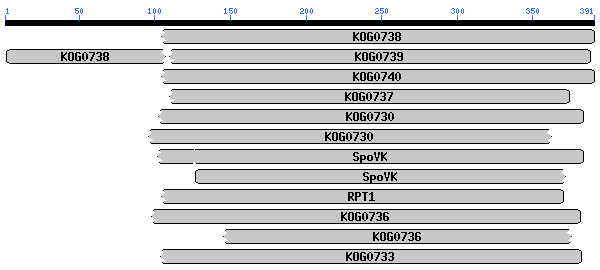
gnl|CDD|18532, KOG0738, KOG0738, KOG0738, AAA+-type ATPase [Posttranslational modification, protein turnover, chaperones] CD-Length = 491 residues, only 58.0% aligned
Score = 505 bits (1302), Expect = 7e-144
Query: 104 PAEHRDDIADLEEAKKLLREAVVLPMWMPDFFKGIRRPWKGVLMVGPPGTGKTMLAKAVA 163 Sbjct: 207 PNIKWDDIAGLHEAKKLLKEAVVLPIWMPEFFKGIRRPWKGVLMVGPPGTGKTLLAKAVA 266 Query: 164 TECGTTFFNVSSSTLTSKYRGESEKLVRLLFEMARFYAPTTIFIDEIDSICSRRGTSDEH 223 Sbjct: 267 TECGTTFFNVSSSTLTSKWRGESEKLVRLLFEMARFYAPSTIFIDEIDSLCSQRGGSSEH 326 Query: 224 EASRRVKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRRRLEKRIYIPLPT 283 Sbjct: 327 EASRRVKSELLVQMDGVQGTLEN---SKVVMVLAATNFPWDIDEALRRRLEKRIYIPLPD 383 Query: 284 AKGRAELLKINLREVELDPDIQLEDIAEKIEGYSGADITNVCRDASLMAMRRRINGLSPE 343 Sbjct: 384 AEARSALIKILLRSVELDDPVNLEDLAERSEGYSGADITNVCREASMMAMRRKIAGLTPR 443 Query: 344 EIRALSKEELQMPVTKGDFELALKKIAKSVSAADLEKYEKWMVEFGSA 391 Sbjct: 444 EIRQLAKEEPKMPVTNEDFEEALRKVRPSVSAADLEKYEKWMDEFGSC 491 gnl|CDD|18532, KOG0738, KOG0738, KOG0738, AAA+-type ATPase [Posttranslational modification, protein turnover, chaperones] CD-Length = 491 residues, only 22.0% aligned
Score = 145 bits (366), Expect = 2e-35
Query: 1 MNLAEICDNAKKGREYALLGNYDSSMVYYQGVMQQIQRHCQSVRDPAIKGKWQQVRQELL 60 Sbjct: 1 MSLAGISENAKLAREYALLGNYDSAGIYYRGLLYLMNRYLVSTGDPYAQGKWSQVEQALT 60 Query: 61 EEYEQVKSIVSTLESFKIDKPPDFPVSCQDEPFRDPAVWPPPVPAEHR 108 Sbjct: 61 EEYELVKQIVRDLRDLKEASTPTLKFSGHDEPPIDPDVWAKPKPVERR 108 gnl|CDD|18533, KOG0739, KOG0739, KOG0739, AAA+-type ATPase [Posttranslational modification, protein turnover, chaperones] CD-Length = 439 residues, only 69.2% aligned
Score = 278 bits (712), Expect = 2e-75
Query: 109 DDIADLEEAKKLLREAVVLPMWMPDFFKGIRRPWKGVLMVGPPGTGKTMLAKAVATECGT 168 Sbjct: 133 SDVAGLEGAKEALKEAVILPIKFPQLFTGKRKPWRGILLYGPPGTGKSYLAKAVATEANS 192 Query: 169 TFFNVSSSTLTSKYRGESEKLVRLLFEMARFYAPTTIFIDEIDSICSRRGTSDEHEASRR 228 Sbjct: 193 TFFSVSSSDLVSKWMGESEKLVKNLFEMARENKPSIIFIDEIDSLCGSRS-ENESEASRR 251 Query: 229 VKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRRRLEKRIYIPLPTAKGRA 288 Sbjct: 252 IKTEFLVQMQGVG----NDNDG--VLVLGATNIPWVLDSAIRRRFEKRIYIPLPEAHARA 305 Query: 289 ELLKINLREVE---LDPDIQLEDIAEKIEGYSGADITNVCRDASLMAMRRRI-------- 337 Sbjct: 306 RMFKLHLGDTPHVLTEQDF--KELARKTEGYSGSDISIVVRDA-LMEPVRKVQSATHFKK 362 Query: 338 --NGLSPEEIRAL--------------------SKEELQMPVTKGDFELALKKIAKSVSA 375 Sbjct: 363 VSGPSNPSEVDDLLTPCSPGDPGAIEMSWMDVPADKLLEPPVTMRDFLKSLSRTKPTVNE 422 Query: 376 ADLEKYEKWMVEFG 389 Sbjct: 423 DDLLKHEKFTEDFG 436 gnl|CDD|18534, KOG0740, KOG0740, KOG0740, AAA+-type ATPase [Posttranslational modification, protein turnover, chaperones] CD-Length = 428 residues, only 65.2% aligned
Score = 258 bits (660), Expect = 2e-69
Query: 104 PAEHRDDIADLEEAKKLLREAVVLPMWMPDFFKGIRRPWKGVLMVGPPGTGKTMLAKAVA 163 Sbjct: 148 RNVGWDDIAGLEDAKQSLKEAVILPLLRPDLFLGLREPVRGLLLFGPPGTGKTMLAKAIA 207 Query: 164 TECGTTFFNVSSSTLTSKYRGESEKLVRLLFEMARFYAPTTIFIDEIDSICSRRGTSDEH 223 Sbjct: 208 TESGATFFNISASSLTSKYVGESEKLVRALFKVARSLQPSVIFIDEIDSLLSKRSDN-EH 266 Query: 224 EASRRVKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRRRLEKRIYIPLPT 283 Sbjct: 267 ESSRRLKTEFLLQFDGK-----NSAPDDRVLVIGATNRPWELDEAARRRFVKRLYIPLPD 321 Query: 284 AKGRAELLKINLREVELDPDI----QLEDIAEKIEGYSGADITNVCRDASLMAMRRRING 339 Sbjct: 322 YETRSLLWK---QLLKEQPNGLSDLDISLLAKVTEGYSGSDITALCKEAAMGPLRELGGT 378 Query: 340 LSPEEIRALSKEELQMPVTKGDFELALKKIAKSVSAADLEKYEKWMVEFGSA 391 Sbjct: 379 TDLEFIDADKI----RPITYPDFKNAFKNIKPSVSLEGLEKYEKWDKEFGSS 426 gnl|CDD|18531, KOG0737, KOG0737, KOG0737, AAA+-type ATPase [Posttranslational modification, protein turnover, chaperones] CD-Length = 386 residues, only 71.0% aligned
Score = 238 bits (609), Expect = 2e-63
Query: 109 DDIADLEEAKKLLREAVVLPMWMPDFFKGIR--RPWKGVLMVGPPGTGKTMLAKAVATEC 166 Sbjct: 92 DDIGGLEEVKDALQELVILPLRRPELFAKGKLLRPPKGILLYGPPGTGKTMLAKAIAKEA 151 Query: 167 GTTFFNVSSSTLTSKYRGESEKLVRLLFEMARFYAPTTIFIDEIDSICSRRGTSDEHEAS 226 Sbjct: 152 GANFINVSVSNLTSKWFGEAQKLVKAVFSLASKLQPSIIFIDEVDSFLGQRRSTD-HEAT 210 Query: 227 RRVKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRRRLEKRIYIPLPTAKG 286 Sbjct: 211 AMMKNEFMALWDGL-----SSKDSERVLVLGATNRPFDLDEAIIRRLPRRFHVGLPDAEQ 265 Query: 287 RAELLKINLREVELDPDIQLEDIAEKIEGYSGADITNVCRDASLMAMRRRIN-GLSPEEI 345 Sbjct: 266 RRKILKVILKKEKLEDDVDLDEIAQMTEGYSGSDLKELCRLAALRPIRELLVSETGLLDL 325 Query: 346 RALSKEELQMPVTKG----------DFELALKKIAKSVSA 375 Sbjct: 326 DKAIADLKPTQAAASSCLLRPLEQEDFPKAINRVSASVAM 365 gnl|CDD|18524, KOG0730, KOG0730, KOG0730, AAA+-type ATPase [Posttranslational modification, protein turnover, chaperones] CD-Length = 693 residues, only 38.0% aligned
Score = 237 bits (605), Expect = 5e-63
Query: 102 PVPAEHRDDIADLEEAKKLLREAVVLPMWMPDFFK--GIRRPWKGVLMVGPPGTGKTMLA 159 Sbjct: 427 EMPNVSWDDIGGLEELKRELQQAVEWPLKHPEKFARFGISPP-KGVLLYGPPGCGKTLLA 485 Query: 160 KAVATECGTTFFNVSSSTLTSKYRGESEKLVRLLFEMARFYAPTTIFIDEIDSICSRRGT 219 Sbjct: 486 KALANEAGMNFLSVKGPELFSKYVGESERAIREVFRKARQVAPCIIFFDEIDALAGSRG- 544 Query: 220 SDEHEASRRVKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRR--RLEKRI 277 Sbjct: 545 GSSSGVTDRVLSQLLTEMDGLEA-------LKNVLVIAATNRPDMIDPALLRPGRLDRII 597 Query: 278 YIPLPTAKGRAELLKINLREVELDPDIQLEDIAEKIEGYSGADITNVCRDASLMAMRRRI 337 Sbjct: 598 YVPLPDLEARLEILKQCAKKMPFSEDVDLEELAQATEGYSGAEIVAVCQEAALLALRESI 657 Query: 338 NGLSPEEIRALSKEELQMPVTKGDFELALKKIAKSVSAADLEKYEKW 384 Sbjct: 658 E--ATE-------------ITWQHFEEALKAVRPSLTSELLEKYEDF 689 gnl|CDD|18524, KOG0730, KOG0730, KOG0730, AAA+-type ATPase [Posttranslational modification, protein turnover, chaperones] CD-Length = 693 residues, only 38.2% aligned
Score = 166 bits (421), Expect = 1e-41
Query: 95 DPAVWPPPVPAEHRDDIADLEEAKKLLREAVVLPMWMPDFFKGIRRPW-KGVLMVGPPGT 153 Sbjct: 170 EPAKREEEELPEVGDDIGGLKRQLSVIRELVELPLRHPALFKSIGIKPPRGLLLYGPPGT 229 Query: 154 GKTMLAKAVATECGTTFFNVSSSTLTSKYRGESEKLVRLLFEMAR-FYAPTTIFIDEIDS 212 Sbjct: 230 GKTFLVRAVANEYGAFLFLINGPELISKFPGETESNLRKAFAEALKFQVPSIIFIDELDA 289 Query: 213 ICSRRGTSDEHEASRRVKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRR- 271 Sbjct: 290 LCPKREGADDVE--SRVVSQLLTLLDGLKPDAK-------VIVLAATNRPDSLDPALRRG 340 Query: 272 RLEKRIYIPLPTAKGRAELLKINLREVELDPDIQLEDIAEKIEGYSGADITNVCRDASLM 331 Sbjct: 341 RFDREVEIGIPGSDGRLDILRVLTKKMNLLSDVDLEDIAVSTHGYVGADLAALCREASLQ 400 Query: 332 AMRRRINGLSPEE--IRALSKEELQMPVTKGDFE 363 Sbjct: 401 ATRRTLEIFQEALMGIRPSALREILVEMPNVSWD 434 gnl|CDD|10337, COG0464, SpoVK, ATPases of the AAA+ class [Posttranslational modification, protein turnover, chaperones] CD-Length = 494 residues, only 52.4% aligned
Score = 226 bits (578), Expect = 6e-60
Query: 101 PPVPAEHRDDIADLEEAKKLLREAVVLPMWMPDFFKGI-RRPWKGVLMVGPPGTGKTMLA 159 Sbjct: 234 FEDEDVTLDDIGGLEEAKEELKEAIETPLKRPELFRKLGLRPPKGVLLYGPPGTGKTLLA 293 Query: 160 KAVATECGTTFFNVSSSTLTSKYRGESEKLVRLLFEMARFYAPTTIFIDEIDSICSRRGT 219 Sbjct: 294 KAVALESRSRFISVKGSELLSKWVGESEKNIRELFEKARKLAPSIIFIDEIDSLASGRGP 353 Query: 220 SDEHEASRRVKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRR--RLEKRI 277 Sbjct: 354 S-EDGSGRRVVGQLLTELDGI-------EKAEGVLVIAATNRPDDLDPALLRPGRFDRLI 405 Query: 278 YIPLPTAKGRAELLKINLREVE--LDPDIQLEDIAEKIEGYSGADITNVCRDASLMAMRR 335 Sbjct: 406 YVPLPDLEERLEIFKIHLRDKKPPLAEDVDLEELAEITEGYSGADIAALVREAALEALR- 464 Query: 336 RINGLSPEEIRALSKEELQMPVTKGDFELALKKIAKSVSAADLEKYEKW 384 Sbjct: 465 ---------------EARRREVTLDDFLDALKKIKPSVT------YEEW 492 gnl|CDD|10337, COG0464, SpoVK, ATPases of the AAA+ class [Posttranslational modification, protein turnover, chaperones] CD-Length = 494 residues, only 46.4% aligned
Score = 117 bits (294), Expect = 4e-27
Query: 126 VLPMWMPD-FFKGIRRPWKGVLMVGPPGTGKTMLAKAVATECGTTFFNVSSSTLTSKYRG 184 Sbjct: 1 ELPLKEPELFKKLGIEPPKGVLLHGPPGTGKTLLARALA-NEGAEFLSINGPEILSKYVG 59 Query: 185 ESEKLVRLLFEMARFYAPTTIFIDEIDSICSRRGTSDEHEASRRVKSELLIQMDGVGGAL 244 Sbjct: 60 ESELRLRELFEEAEKLAPSIIFIDEIDALAPKR-SSDQGEVERRVVAQLLALMDGLKRGQ 118 Query: 245 ENDDPSKMVMVLAATNFPWDIDEALRR--RLEKRIYIPLPTAKGRAELLKINLREVELDP 302 Sbjct: 119 --------VIVIGATNRPDGLDPAKRRPGRFDREIEVNLPDEAGRLEILQIHTRLMFLGP 170 Query: 303 DIQLEDIAEKIEGYSGADITNVCRDASLMAMRRRINGLSPEEIRALSKEELQMPVTKGDF 362 Sbjct: 171 PGTGKTLAARTVGKSGADLGALAKEAALRE-LRRAIDLVGEYIG----------VTEDDF 219 Query: 363 ELALKKIAKS 372 Sbjct: 220 EEALKKVLPS 229 gnl|CDD|10940, COG1222, RPT1, ATP-dependent 26S proteasome regulatory subunit [Posttranslational modification, protein turnover, chaperones] CD-Length = 406 residues, only 61.3% aligned
Score = 220 bits (563), Expect = 4e-58
Query: 104 PAEHRDDIADLEEAKKLLREAVVLPMWMPDFFK--GIRRPWKGVLMVGPPGTGKTMLAKA 161 Sbjct: 146 PDVTYEDIGGLDEQIQEIREVVELPLKNPELFEELGIDPP-KGVLLYGPPGTGKTLLAKA 204 Query: 162 VATECGTTFFNVSSSTLTSKYRGESEKLVRLLFEMARFYAPTTIFIDEIDSICSRR---G 218 Sbjct: 205 VANQTDATFIRVVGSELVQKYIGEGARLVRELFELAREKAPSIIFIDEIDAIGAKRFDSG 264 Query: 219 TSDEHEASRRVKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRR--RLEKR 276 Sbjct: 265 TSGDREV-QRTMLELLNQLDGF-------DPRGNVKVIMATNRPDILDPALLRPGRFDRK 316 Query: 277 IYIPLPTAKGRAELLKINLREVELDPDIQLEDIAEKIEGYSGADITNVCRDASLMAMRRR 336 Sbjct: 317 IEFPLPDEEGRAEILKIHTRKMNLADDVDLELLARLTEGFSGADLKAICTEAGMFAIRER 376 Query: 337 INGLSPEEIRALSKEELQMPVTKGDFELALKKIAK 371 Sbjct: 377 -----------------RDEVTMEDFLKAVEKVVK 394 gnl|CDD|18530, KOG0736, KOG0736, KOG0736, Peroxisome assembly factor 2 containing the AAA+-type ATPase domain [Posttranslational modification, protein turnover, chaperones] CD-Length = 953 residues, only 30.0% aligned
Score = 220 bits (562), Expect = 5e-58
Query: 97 AVWPPPVPAEHRDDIADLEEAKKLLREAVVLPMWMPDFFKGIRRPWKGVLMVGPPGTGKT 156 Sbjct: 660 AIGAPKIPNVSWDDVGGLEEVKTEILDTIQLPLKHPELFSSGLRKRSGILLYGPPGTGKT 719 Query: 157 MLAKAVATECGTTFFNVSSSTLTSKYRGESEKLVRLLFEMARFYAPTTIFIDEIDSICSR 216 Sbjct: 720 LLAKAVATECSLNFLSVKGPELLNMYVGQSEENVREVFERARSAAPCVIFFDELDSLAPN 779 Query: 217 RGTS-DEHEASRRVKSELLIQMDGVGGALENDDPSKMVMVLAATNFPWDIDEALRR--RL 273 Sbjct: 780 RGRSGDSGGVMDRVVSQLLAELDGL-----SDSSSQDVFVIGATNRPDLLDPALLRPGRF 834 Query: 274 EKRIYIPLP-TAKGRAELLKINLREVELDPDIQLEDIAEKI-EGYSGADITNVCRDASLM 331 Sbjct: 835 DKLVYVGPNEDAESKLRVLEALTRKFKLDEDVDLVEIAKKCPPNMTGADLYSLCSDAMLA 894 Query: 332 AMRRRINGLSPEEIRALSKEELQM | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||