ANALYSIS OF THE OUTFILE
Once the outfile was obtained, we contrasted our expectations in relation with the content of this file. We evaluated the following parameters:
This command calculates how many times this symbol "## date" is found within the outfile. We chose this symbol because it is included in every prediction that Geneid makes. The returned value was 60,770 so we concluded that there was a prediction for each sequence of cDNA.
This command calculates how many times this symbol ">" is found within the outfile. This symbol precedes the ID of each sequence predicted in FASTA format. Contrary to our belief, it turned out that the number of predicted proteins was lower than expected (43,480).
To check it we used the following Unix command:
We obtained these results:
- Forward: 37,758
Reverse: 5,722 (*)
(*) We observed some cDNAs reverse sequences carrying a good score in the outfile, which means that not all the sequences were introduced in forward, as the article ensured.
To check which proportion of the 5,722 sequences had a significant score, we fixed a threshold of score > 10:
The result provided by this Unix command was: 369 sequences of cDNA.
When we observed that a large number of sequences in reverse with a good score had been obtained, we decided to increase the threshold value to have an idea of the amount of cDNA sequences carrying a score higher than 20:
The result obtained with the last Unix command was 109 cDNA sequences.
As soon as we saw the last results, we wanted to know if the presence of high score values were due to bad predictions or to the fact that some of the sequences had been introduced in reverse. It was likely to think that those sequences carrying a higher score values corresponded to true proteins.
To validate this, we performed a pBLAST search using some of the predicted proteins provided by Geneid (score > 20).
We figured out that all these proteins checked were real proteins, e.g. the sequence with a score > 50 corresponded to a kinase well characterized in mouse.
The result was:
- Single: 10,139
First: 10,322
Internal: 17,993
Terminal: 13,327
In theory, we would expect to have only a single exon for each predicted protein because we are treating with cDNA sequences which are monocistronic in eukaryota organisms. The reason of these results could be due to the fact that Geneid predicts donnor and acceptor sites which are not real.
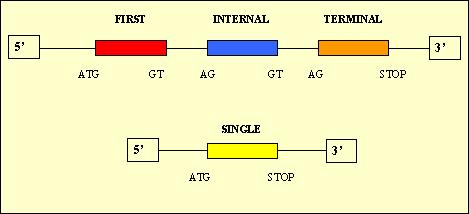
Moreover, it is supposed to be only one exon for each predicted protein, but we verified that it did not occur:
This Unix command provided us the total number of exons predicted (51,781) which should coincide with the number of predicted proteins(43,480).
As they are different, it is indicative that for some cDNAs it is predicted more than one exon.
IMPROVING OUR PREDICTIONS
Our aim was to improve the Geneid predictions by modifying the parameter file. For this, we created a gene model in which we inactivated the intronic connections because we are treating with cDNAs which have no introns. As well, we forced intergenic connections to pedict only single exons, inactivating the other connections between those non-single exons.
Next, we show the modified gene model:
|
# # |
# # |
50:4000 50:4000 |
|
Single+ Single+ Single- # # # |
Single+ Single- Single+ # # # |
500:Infinity 500:Infinity 500:Infinity 500:Infinity 500:Infinity 500:Infinity |
After making predictions, we worked the sequences out obtaining the number of single exons in forward and in reverse. Moreover, we also counted how many predictions had been made by Geneid.
The result was:
- Single: 31,592 (equal to the number of predictions)
Forward: 30,088
Reverse: 1,504
At this point, we decided to work only with those predictions in forward because of the biological restrictions, as we have commented before.
THE EVIDENCES
The next step consisted of extracting those cDNAs containing IRE structures from the input file (60,770 sequences). It was possible thanks to the file provided by Ana Igea & Iris Uribesalgo which included the IDs of those cDNA sequences (594) showing IRE evidences (601). The final objective was to use a gff file with the predicted IREs as external evidence in the prediction performed by Geneid on these 594 cDNA. We created a new version of the Predictor2.0.pl to perform this task.
The protocol that we followed was:
We observed that there were 594 cDNA sequences. Because of some cDNAs matched the same IREs IDs, the number of cDNA sequences is lower than 601.