Distribució de SNPs en el Genoma Humà
DISTRIBUCIÓ D'SNPs EN EL GENOMA HUMÀ
Laura Corominas i Carla Carbó
INTRODUCCIÓ I OBJECTIUS
En els darrers anys els estudis relacionats amb el
genoma humà han centrat bona part dels esforços de la comunitat científica. Amb la recent seqüenciació del genoma humà s'ha generat una allau d'informació molt gran però ara ens calen diferents eines per tal de poder-la assimilar. Això ha donat lloc a l'aparició d'un gran nombre d'estudis en els diferents camps relacionats amb la recerca i la medicina. En aquest moment ens trobem als inicis de l'exploració d'aquesta informació que es preveu que, en un futur més o menys pròxim, la podem aplicar. Aquest enorme potencial es veurà especialment reflectit en múltiples avenços en el diagnòstic, tractament i descobriment de malalties.
Actualment s'estan intentant identificar polimorfismes entre la població humana, entre ells s'hi troben els SNPs, que són variacions d'un nucleòtid presents en més d'un 1% de la població.
L'objectiu d'aquest treball és estudiar la distribució dels SNPs en un subconjunt de gens del genoma humà. Ens interessa conèixer la proporció d'SNPs en cadascuna de les regions del genoma: exons, introns, UTRs, llocs d'splicing i regions intergèniques. A partir dels resultats obtinguts intentarem respondre algunes preguntes que se'ns plantegen:
- La distribució d'SNPs és homogènia en el genoma?
- Quines regions accepten més variació nucleotídica?
- Existeixen diferències entre cromosomes pel que fa a la presència d'SNPs?
INICI
MATERIALS I MÈTODES
Els programes han estat creats amb el llenguatge de programació
Perl en el sitema operatiu
Linux. Pel tractament d'algunes dades s'ha utilitzat el progama
Excel del sistema operatiu Microsoft Windows. Per elaborar la pàgina web (amb
codi HTML) s'ha utlitzat l'editor de text emacs del Linux.
MATERIALS
Per fer el nostre treball hem utilitzat els fitxers locatlitzats en el UCSC browser (versió del 14 de novembre del 2002): snpNih.txt (conté informació sobre la localització dels SNPs en el genoma humà) i refGene.txt (que conté la localització exònica d'aproximadament 18000 gens humans).
Conèixer el format dels dos fitxers ha estat essencial a l'hora de treballar amb ells:
- El fitxer dels SNPs (snpNih.txt) és un fitxer on a cada fila hi ha descrit un SNP i, 4 columnes on, a la primera hi tenim el cromosoma on es troba l'SNP, la segona i la tercera corresponen a la posició d'inici i terminació de l'SNP, i la quarta és un número identificador. D'aquest fitxer les columnes que ens han interessat han estat la primera i la tercera.
- El fitxer dels gens humans (refGene.txt) té una estructura semblant. A cada fila hi ha un gen i a les 9 columnes la informació que tenim, per ordre, és la següent: identificador, cromosoma, forward/reverse strand (+/-), inici del trasnscrit, fi del transcrit, inici del CDS, fi del CDS, número d'exons, inici de tots els exons i fi de tots els exons.
MÈTODES
Obtenció dels fitxers
Baixar els dos fitxers des de la pàgina web de l'assignatura on hi tenim el link d'accés. Descomprimir-los i guardar-los sota el mateix nom. Per fer-ho:
$gzip -d refGene.txt
$gzip -d snpNih.txt
Programa
Fer un programa que ens compta quants SNPs hi ha en les següents regions (per cada gen):
- 5'UTR
- 3'UTR
- UTR total
- INTRONS
- EXONS
- CDS
- DONORS
- ACCEPTORS
- SPLICING
- TOTAL
Execució del programa i obtenció dels resultats.
El format de sortida dels resultats és un fitxer (solucio.txt) amb columnes. A la primera columna hi ha l'identificador del gen i a les següents hi trobem el valor associat (quantitat d'SNPs) a: UTR3, UTR5, UTRtotal, INTRONS, EXONS, CDS, DONORS, ACCEPTORS, SPLICING i TOTAL d'SNPs del gen.
$./programa.txt refGene.txt snpNih.txt > solucio.txt
Tractament de les dades
A partir de diferents comandes des del shell realitzem algunes operacions i modifiquem el fitxer solucio.txt per tal de poder analitzar els resulats.
a) Suma dels SNPs totals en les diferents regions i per cromosomes:
Sumes totals:
- Posar cada columna en un fitxer:
$cut -f2 solucio.txt > UTR3.txt
$cut -f3 solucio.txt > UTR5.txt
$cut -f11 solucio.txt > TOTAL.txt
...
D'aquesta manera tenim tots els SNPs que es troben en les diferents regions dins un fixer amb una sola columna.
La suma dels valors que conté el fitxer TOTAL.txt és el total d'SNPs que ha calculat el programa. Aquesta dada serà utilitzada per fer les proporcions i distribucions de la majoria de gràfics.
- Fer un programa que ens sumi columnes (suma_SNP.pl).
- Amb els resultats fer els gràfics per poder analitzar posteriorment.
Sumes per cromosomes:
- Posem tots els gens d'un cromosoma en un fitxer (un per cromosoma). Llavors seleccionem les columnes que contenen la informació de UTRs, exons, introns, regions d'splicing i totals. Ho fem (exemple pel cromosoma 1):
$egrep '(chr1\b)' solucio.txt > chr1.txt
$cut -f4 chr1.txt > utrc1.txt
$cut -f5 chr1.txt > exoc1.txt
$cut -f6 chr1.txt > intc1.txt
$cut -f10 chr1.txt > splc1.txt
$cut -f11 chr1.txt > totalc1.txt
Així tenim un fitxer amb els SNPs del cromosoma 1 que es troben en UTRs, un amb els que es troben en exons, un amb els d'introns, un amb els de regions d'splicing i un altre am els totals; i això ho tenim per tots els cromosomes.
- Correm el programa suma_SNP.pl per cada fitxer.
- Els resultats els passem a Excel i en fem els gràfics corresponents.
b) Estudiar la distribució dels SNPs:
La distribució l'estudiem a nivell de genoma i a nivell de cromosomes.
Genoma:
Fem un programa que ens calcula la distribució (distribucions.pl), és a dir, el número de gens que tenen un rang determinat d'SNPs.
Al programa li passem el fitxer del total d'SNPs dels gens (TOTAL.txt).
$./distribucions.pl TOTAL.txt > distgenoma.txt
El resultat el tractem a l'Excel on en fem un gràfic.
Cromosomes:
Fem dos tipus de tractament de dades:
- $egrep '(chr1)' solucio.txt > CHR1.txt
$cut -f11 CHR1.txt > C1.txt
$./suma_snp.pl C1.txt
Amb aquestes operacions obtenim el total d'SNPs que té cada cromosoma.
A partir d'aquestes dades construïm una taula i fem un gràfic.
- Per cada cromosoma comptem el número de gens que tenen un rang determinat d'SNPs.
Agafem els fitxers C1.txt, C2.txt, ... (obtinguts anteriorment amb les comandes egrep i cut) i correm el programa distribucions.pl:
$./distribucions.pl C1.txt
$./distribucions.pl C2.txt
...
A partir dels resultats obtinguts fem un perfil de distribucions per cada cromosoma (utilitzant l'Excel).
A més a més, utilitzem diferents comandes del shell per tenir altres dades:
- Comptar el número de gens i el número d'SNPs totals dels dos fitxers.
$wc refGene.txt
$wc snpNih.txt
La comanda wc ens permet comptar el total de gens i d'SNPs perquè a cada línia dels fixters hi ha un gen i un SNP, respectivament.
- Comptar el número de gens que hi ha per cromosoma.
$wc Chr1.txt
$wc Chr2.txt
...
Fem el wc dels fitxers que tenen els gens de cada cromosma.
Resultats
Es mostren més endavant.
INICI
PROGRAMES
Per fer l'anàlisi de les dades hem fet tres programes: el que ens compta quants SNPs hi ha en cada regió (introns, exons, ...) per gen (programa.pl), el que ens suma els SNPs per regions (suma_SNP.pl) i el que ens calcula la distribució d'SNPs en els gens.
programa.pl
L'estructura del programa és la següent:
- Declaració de variables. Són variables generals per tot el programa (en declarem moltes més dins cada funció però només tenen validesa mentre s'executa la funció).
- Main loop, que consta de 5 funcions:
- Obrir el fitxer refseq (és, de fet, el fitxer refGene.txt). Creació i definició del hash %refgene.
- Obrir el fitxer SNPs (és el fitxer snpNih.txt). Creació i definició del hash %snpNih.
- Ordenar l'array d'SNPs. Ordenem els SNPs de cada cromosoma per posició, per tal de fer més àgils les operacions.
- Tractament de les dades. Utilitzant els hashes creats anteriorment i, a partir d'una sèrie de composicions iteratives, el programa ens compta els SNPs que es troben en cadascuna de les regions a analitzar, per cada gen. Creació d'un tercer hash %resultats.
- Imprimir els resultats.
suma_SNP.pl
Aquest programa el que fa és posar números d'una columna dins un vector i sumar-los (l'hem dissenyat així ja que els nostres fitxers d'entrada tenen aquest format).
El programa realitza tres operacions:
- Càlcul dels SNPs absoluts.
- Càlcul de la freqüència relativa respecte tot el genoma. Utilitzem tots els SNPs del fitxer original
- Càlcul de la freqüència relativa respecte les regions gèniques. Utilitzem només els SNPs que el programa.pl ens ha localitzat.
distribucions.pl
Aquest programa ens calcula la distribució dels SNPs en els gens, és a dir, quants gens tenen un rang determinat d'SNPs, per exemple: entre 0 i 5, entre 5 i 10, ... i fins a més de 700.
Els rangs del programa els hem decidit després d'observar els resultats obtinguts. Per aquest motiu, hem cregut necessari fer rangs de mida variable. D'aquesta forma evitem que s'acumulin massa dades en el mateix rang i obtenim una descripció més informativa.
INICI
RESULTATS I ANÀLISI
L'objectiu principal d'aquest treball era conèixer la distribució dels SNPs en les diferents regions del genoma: UTRs, exons, introns, llocs d'splicing i regions intergèniques.
El resultat obtingut després de còrrer el programa es troba en
solucio.txt.
La presentació dels resultats està estructurada de la següent manera:
A) ANÀLISI GLOBAL DEL GENOMA
A.1) PROPORCIÓ D'SNPs EN EL GENOMA
En primer lloc, construïm una taula on observem els SNPs totals per a cada regió, i fem el gràfic de la proporció d'SNPs respecte tot el genoma. És a dir, hem calculat el % d'SNPs de cada regió segons el total d'SNPs que hi ha descrits en el genoma humà (fitxer snpNih.txt): 1.073.363 SNPs.
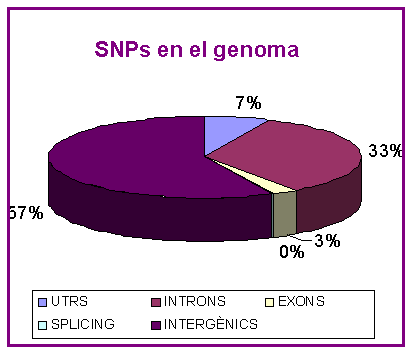
El programa situava els SNPs tenint en compte les posicions de principi i final de cada transcrit; per aquest motiu no ens comptava els SNPs de regions intergèniques. Aquest valor serà la diferència entre els SNPs del fitxer snpNih.txt i els SNPs que el programa ha trobat en les regions gèniques: 1.073.363 - 459.675 = 613.688
Observem que la majoria d'SNPs es troben precisament en aquestes regions entre gens (57%). El 43% restant es localitza dins els gens que estem analitzant. Els SNPs trobats en introns tenen també un elevat valor i representen el 33% de tot el genoma. Els SNPs en UTRs, exons i llocs d'splicing representen el 7%, 3% i 0%, respectivament.
El 0% d'SNPs obtingut en el llocs d'splicing no significa que no hi hagi polimorfismes en aquestes posicions, sinó que la variació és molt petita si la comparem amb la variació de tot el genoma. De fet, aquests pocs canvis de nucleòtid que ara podrien semblar insignificants esdevindran un dels punts més interessants a analitzar.
B) ANÀLISI DE LES REGIONS GÈNIQUES
B.1) PROPORCIÓ D'SNPs EN LES REGIONS GÈNIQUES
Ara construïm una altra taula i un gràfic on també observarem els SNPs de cada regió, però sense tenir en compte els que es troben a regions intergèniques. És a dir, les freqüències relatives estan calculades respecte el total d'SNPs que es troben en regions gèniques: 459.675 SNPs. A partir d'aquí tots els gràfics estan calculats respecte aquesta dada.

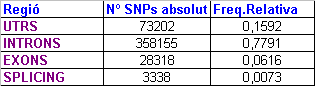
Com ja hem vist en el gràfic anterior, la majoria d'SNPs que es troben en els gens cauen en regions intròniques, representant el 77% del total d'SNPs en regions gèniques. Els exons són el 6%, els UTRs el 16% i els llocs d'splicing l'1%.
B.1.1) UTR5' I UTR3'
En veure que hi havia un nombre important d'SNPs en els UTRs ens vam plantejar si podrien existir diferències entre l'UTR 5' i l'UTR 3'. Per aquest motiu, vam modificar el programa original de forma que ens comptés per separat els SNPs que hi havia en els dos extrems de cadascun dels gens. A continuació mostrem la taula i el gràfic que recullen aquesta informació:
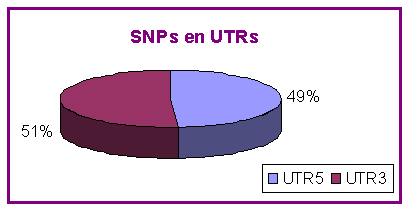
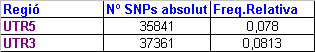
La distribució d'SNPs és aproximadament la meitat en cada UTR. La diferència és mínima.
B.1.2) DONORS I ACCEPTORS
Pel que fa als llocs d'splicing, el programa està pensat per tal de diferenciar els SNPs que cauen en Donors i en Acceptors. Com que l'splicing és un procés essencial a l'hora de processar l'mRNA, aquesta informació pot ser molt important:
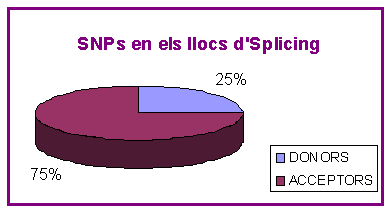

En el gràfic podem observar que els SNPs són més freqüents en en les posicions d'acceptors (75%) que en les posicions de donors (25%).
B.2) DISTRIBUCIÓ D'SNPs EN ELS GENS
El programa ens calculava el total d'SNPs identificats en cada gen. Per tenir una visió general de com és la variabilitat en les regions gèniques, hem fet la següent gràfica de distribució:
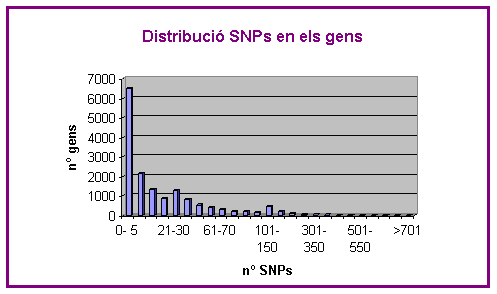
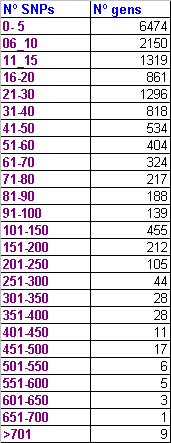
Per fer el gràfic hem agafat els valors totals obtinguts (fitxer TOTAL.txt) amb el programa.pl i hem corregut el programa de distribucions.pl.
Observem que molts gens (41,4%) tenen entre 0 i 5 SNPs (primer pic). També hi ha bastants gens (13,7%) que contenen de 6 a 40 SNPs, dins aquest rang es troba un segon pic entre 21 i 30. El número de gens que contenen entre 41 i 250 SNPs és menor, però també significatiu, i hi trobem un tercer pic entre 101 i 150. A partir d'aquí, a mesura que augmenta el nombre de SNPs disminueix el nombre de gens que els contenen.
C) ANÀLISI PER CROMOSOMES
A més d'analitzar la informació per tot el genoma en general, ens interessa fer-ho per cromosomes. A continuació mostrarem una sèrie de gràfics on la informació de gens i SNPs ha estat agrupada segons els cromosomes.
C.1) PROPORCIÓ D'SNPs EN CADA CROMOSOMA
En primer lloc, volíem saber la proporció d'SNPs que hi ha en cadascun dels cromosomes. Per realitzar aquest gràfic vam usar els resultats totals d'SNPs de cada gen calculats amb programa.pl, vam agrupar-los per cromosomes (des del shell) i vam sumar-los amb suma_SNP.pl:
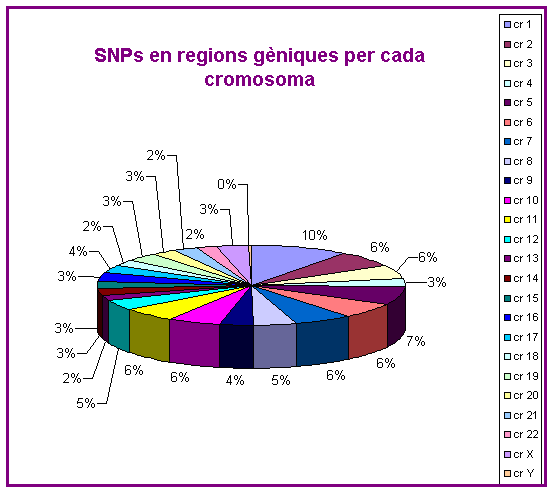
Podem veure que el cromosoma que té més SNPs és el cromosoma 1 (10%). El cromosoma 5 en té un 7%, els cromosomes 2, 3, 6, 7, 10 i 11, tenen un 6% d'SNPs cadascun; els cromosomes 8 i 12, un 5%; els cromosomes 9 i 17, un 4%; els cromosomes 4, 14, 15, 16, 19, 20 i X, un 3% i els cromosomes 13, 18, 21 i 22, un 2%. El cromosoma Y té un 0% però no significa que no tingui SNPs, sinó que te un valor molt petit respecte a la resta de cromosomes.
Per analitzar aquestes proporcions cal tenir en compte que els cromosomes són diferents pel que fa a tamany i número de gens.
La següent taula mostra el número de gens i d'SNPs que tenim per cada cromosoma:
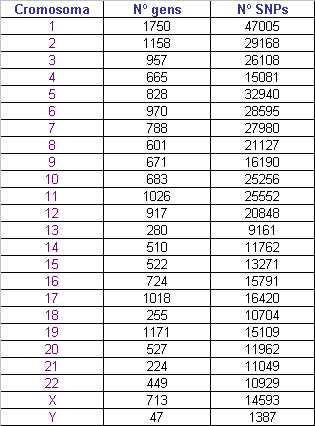
En aquesta taula podem veure que hi ha una distribució desigual tant en número de gens com en quantitat d'SNPs. Això ho haurem de tenir en compte a l'hora d'extreure'n conclusions.
C.2) PROPORCIÓ D'SNPs EN REGIONS GÈNIQUES (PER CORMOSOMES)
En els primers gràfics de sectors mostràvem com estaven distribuits tots els SNPs en les regions gèniques: exons, introns, UTRs, llocs d'splicing... Ara volem mirar si hi ha diferències entre cromosomes. Per això mostrem la taula que conté els resultats en valor absolut i fem el gràfic:
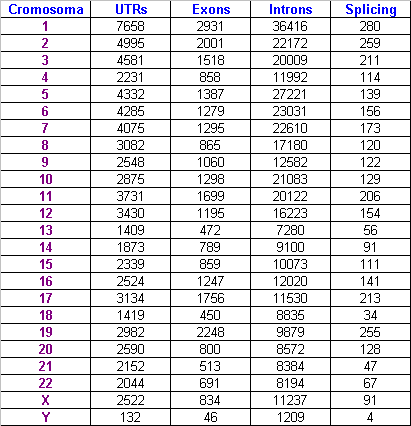
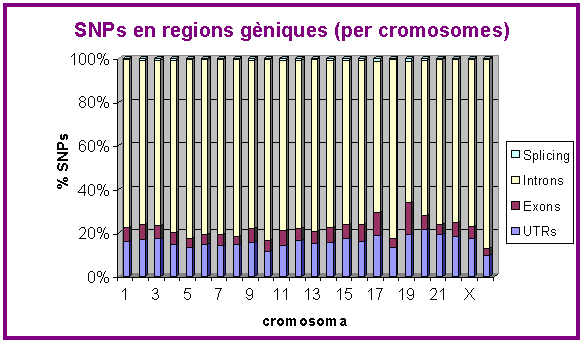
En aquest gràfic veiem que la distribució d'SNPs en regions gèniques és similar en tots els cromosomes. La proporció d'SNPs en introns, exons, UTRs i llocs d'splicing es manté en cadascun d'ells.
C.3) DISTRIBCUIÓ D'SNPs EN ELS CROMOSOMES
Abans hem vist la distribució d'SNPs en els gens. Ara, volem fer una nova distribució dels SNPs respecte els cromosomes. Ens interessa saber quants cromosomes tenen un nombre d'SNPs determinat.
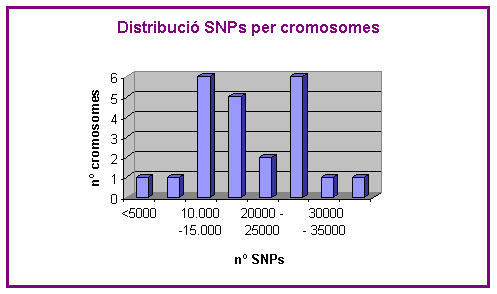
Trobem:
- 1 cromosoma que te menys de 5.000 SNPs
- 2 cromosomes que tenen 5 - 10.000 SNPs
- 6 cromosomes que tenen 10 - 15.000 SNPs
- 5 cromosomes que tenen 15 - 20.000 SNPs
- 2 cromosomes que tenen 20 - 25.000 SNPs
- 6 cromosomes que tenen 25 - 30.000 SNPs
- 1 cromosoma que te 30 - 35.000 SNPs
- 1 cromosoma que te més de 35.000 SNPs
Així doncs, hem fet dos tipus de distribucions:
- Quants gens tenen un determinat número d'SNPs.
- Quants cromosomes tenen un determinat número d'SNPs.
C.4) DISTRIBCUIÓ D'SNPs EN ELS GENS (PER CROMOSOMA)
Volíem saber també, si el perfil de distribució dels SNPs en els gens es mantenia en tots els cromosomes. Per això vam mirar la distribució dels SNPs en els gens però per casdascun dels cromosomes, elaborant el perfil de cada un d'ells. Els resultats obtinguts mostraven un perfil similar en tots els cromosomes, que corresponia amb el perfil que ja havíem observat quan ho fèiem pels gens de tot el genoma. Degut a l'elevada similitud d'aquests perfils, mostrem només el global de tot el genoma.
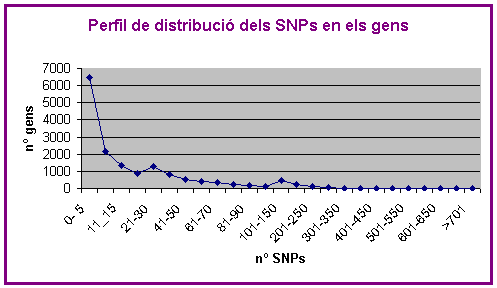
Podem observar que en aquest perfil apareixen els tres pics comentats anteriorment (0-5, 21-30, 101-150).
INICI
CONCLUSIONS
En primer lloc extraurem les conclusions dels gràfics i taules mostrats en l'apartat de resultats. S'estructuren segons 3 grans blocs:
A) PROPORCIONS D'SNPs EN INTRONS, EXONS, UTRs, SPLICING I REGIONS INTERGÈNIQUES
B) PROPORCIÓ D'SNPs EN ELS CROMOSOMES
C) DISTRIBUCIÓ D'SNPs
A) PROPORCIONS D'SNPs EN INTRONS, EXONS, UTRs, SPLICING I REGIONS INTERGÈNIQUES
(Ens referim als apartats A i B dels resultats)
En primer lloc volem destacar el gran nombre d'SNPs que es troben en regions intergèniques en comparació amb la quantitat presents dins els gens. La multitud d'SNPs localitzats en aquestes regions, i també en les regions intròniques s'explica pel fet que es tracta de zones que no
es traduiran a proteïna. Aquestes zones es caracteritzen per tenir
menor pressió selectiva i, per tant, acceptar més variació.
En les regions d'exons, en canvi, trobem justament el contrari. Observem que en aquestes
regions hi ha pocs canvis nucleotídics ja que codifiquen per
proteïnes. Aquestes zones es caracteritzen per tenir una forta
pressió selectiva i no tolerar gaire variació.
La presència d'un SNP en els exons serà més o menys
tolerada segons si aquest comporta o no un canvi d'aminoàcid. La selecció
Natural actuarà sobretot en aquells casos on hi hagi modificació
d'aminoàcid, ja que aquest fet pot tenir repercussions a nivell de
proteïna. Per exemple, pot alterar la funció proteïca,
l'estructura, crear impediments estèrics, resultar en proteïnes truncades, ... Tot això no es donaria si el canvi de nucleòtid fos sinònim.
Les regions d'UTRs contenen major quantitat d'SNPs que els exons
però molt menor que els introns. D'una banda, són regions que no es
traduiran a proteïna, per tant accepten certa variació.
Però per altra banda, són llocs essencials per la
transcripció i traducció dels gens. Els senyals més
importants que s'hi troben són:
- A l'UTR 5':
- CAP (G metilat) que protegeix de la degradació del mRNA una
vegada s'han transcrit uns 30 nucleòtids.
- Dominis on s'uniran factors de transcripció.
- Promotors que regularan la transcripció dels gens.
- TATA box (s'hi uneixen els factors de transcripció i més
tard s'hi unirà la RNApol II).
- A l'UTR 3':
- Cua poliA, que és el senyal de terminació de transcripció.
- Senyals de localització intracitoplasmàtica del mRNA.
Volíem saber el grau de conservació entre l'UTR5' i
l'UTR3' mesurant-ne la quantitat d'SNPs (apartat B.1.1 dels resultats). Hem vist que els dos UTRs
contenen la mateixa proporció d'SNPs. Així doncs podem dir,
que no hi ha diferències significatives entre ells, pel que fa a aquest paràmetre (proporció d'SNPs). Semblaria doncs, que la pressió selectiva és igual en els dos extrems.
Les zones on s'observa una menor quantitat d'SNPs són les regions
d'splicing. L'splicing és el processament del pre-mRNA a mRNA madur, el
qual es traduirà a proteïna. L'alta conservació
d'aquestes posicions ens indica que hi ha una forta pressió
selectiva. Les variacions d'aquests nucleòtids són poc
acceptats ja que interferirien en el processament del mRNA donant lloc
a proteïnes truncades i/o no funcionals.
Hem estudiat si hi havia diferències entre els donors i els acceptors - les dues posicions d'splicing (apartat B.1.2 dels resultats). Hem vist que els acceptors
contenen tres vegades més SNPs que els donors (75% vs 25% d'SNPs en llocs d'splicing).
Una de les possibles explicacions és que definim l'acceptor amb un nombre
major de bases i, per tant, hi ha més probabilitat que hi hagi
algun SNP.
S'han observat les mateixes proporcions d'SNPs en cadascuna d'aquestes
regions (exons, introns, UTRs, splicing) tant a nivell de genoma (resultats B.1) com a nivell de cromosoma (resultats C.2). Per tant, la
Selecció, actua de la mateixa manera en tots els cromosomes.
Les petites diferències entre cromosomes es deuen a la diferent
longitud d'aquests i al nombre de gens que contenen.
B) PROPORCIÓ D'SNPs EN ELS CROMOSOMES
(Ens referim a l'apartat C.1 dels resultats)
El gràfic mostrat s'ha d'analitzar
tenint en compte la taula que hi adjuntem, on hi ha el nombre de gens per cromosoma, ja que la
quantitat d'SNPs estàrà relacionada amb la quantitat de gens
descrits per cada cromosoma.
Observem que el cromosoma amb major proporció d'SNPs és
també el que té un major nombre de gens descrits (cr 1).
De la mateixa manera, el cromosoma Y és el que té menys gens descrits
i menor % d'SNPs. Pel que fa a la resta de cromosomes, la relació
entre aquests dos paràmetres es manté en la majoria d'ells.
No obstant, hi ha alguns casos on això no es dóna
La relació seria encara molt millor si es tingués en compte la
longitud de cadascun dels gens (i no només el nombre de gens). D'aquesta manera es veuria amb més claredat si el nombre d'SNPs trobats en cada cromosoma és proporcional a la longitud de les regions gèniques. Seria d'espererar que això fos així, si assumim que la Selecció Natural actua amb la mateixa força en cada cromosoma.
La diferència en quantitat d'SNPs entre els cromosomes també
pot ser explicada perquè:
- No s'hagi dedicat el mateix esforç per localitzar els SNPs
dels diferents cromosomes.
- Treballem amb un subconjunt dels gens humans.
Aquests dos aspectes poden desviar les proporcions d'SNPs que localitzem en els cromosomes humans.
C) DISTRIBUCIÓ D'SNPs:
Mirant la distribució d'SNPs en el gens (resultats de l'apartat B.2) podem concloure que bastants dels gens estudiats accepten poca quantitat de variació.
Trobem més de la meitat dels gens (55,1%) que tenen entre 0 i 10 SNPs, i un 75% d'aquests tenen només entre 0 i 5 SNPs. L'altra meitat dels gens es troben distribuits en els rangs de més quantitat d'SNPs. El 99% dels gens es troben per sota els 250 SNPs. A més, s'observa que a mesura que augmenta el rang d'SNPs disminueix el nombre de gens que els contenen.
Això confirmaria que la Selecció Natural actua sobre els gens, impedint un nombre elevat d'SNPs en la majoria d'ells.
Mirant el perfil decreixent d'aquesta distribució identifiquem 3 pics diferents:
- el primer entre 0 i 5 SNPs (en aquest primer rang s'inclouen el 41,4% dels gens).
- el segon entre 21 i 30 SNPs (fins aquesta posició tenim el 77,3% dels gens).
- el tercer entre 101 i 150 SNPs (fins aquesta posició trobem el 97% dels gens).
Aquests 3 pics també els vam observar en fer el perfil de distribució per cadascun dels cromosomes (apartat C.4 dels resultats). Caldria pensar doncs, que són rangs importants per entendre l'acceptació d'SNPs en tot el genoma.
En realitat, però, només el primer pic és informatiu, ja que inclou un nombre significatiu de gens.
En canvi, els altres dos pics no serien informatius perquè contenen poca quantitat de gens (8,2% el segon pic i 2,9% el tercer pic). A més, pensem que són artefactes dels rangs seleccionats. Per fer els rangs d'SNPs, vam fer 2 canvis en l'amplitud d'aquests. El segon i tercer pic coincideixen justament en aquests canvis d'escala, i això ens fa pensar que no són pics reals, sinó artefactes. En aquests punts s'acumulen massa SNPs, i segurament caldria haver fet un canvi d'amplitud menor, per tal d'obtenir una gràfica decreixent i sense falsos pics.
Així doncs, podem concloure que la relació entre la quantitat d'SNPs i la quantitat de gens que els contenen és inversament proporcional.
Per últim analitzarem els resultats de l'apartat C.3 que mostren la distribució d'SNPs respecte els cromosomes.
La majoria de cromosomes es troben en els rangs migs d'SNPs: 19 dels 24 cromosomes estudiats tenen un rang d'SNPs entre 10.000 i 30.000. Tot i que hi ha una zona amb només dos cromosomes (de 20 a 25.000 SNPs). En els rangs dels extrems, en canvi, trobem pocs cromosomes: 3 cromosomes tenen menys de 10.000 SNPs i 2 cromosomescontenen un nombre d'SNPs superior a 30.000.
Amb aquestes dades, podem concloure que:
a) D'una banda, que la majoria de cromosomes es troben sota una pressió similar.
b) D'altra banda, que aquesta pressió es troba en rangs intermitjos, és a dir, que no s'accepten molts SNPs, però sí una quantitat considerable (almenys uns 10.000 per cromosoma). Aquest equilibri d'acceptació de canvis pot ser el resultat de que algunes regions gèniques accepten molts SNPs (com els introns) i altres no gaires (com exons, donors i acceptors).
Com a conclusió final, podem dir que la Selecció Natural acuta amb diferent intensitat segons les regions tinguin més o menys significat biològic. Les regions amb menys SNPs corresponen a les relacionades amb el processament i la síntesi de proteïnes, i les regions amb més SNPs són les que tenen menys implicació funcional a nivell de proteïna.
L'estudi de les regions exòniques i d'splicing té rellevància en la recerca biomèdica, ja que algunes de les proteïnes no funcionals provocades per SNPs poden
ser la causa de diferents malalties. Per això, identificar els polimorfismes responsables d'aquestes malalties
permetrà obrir noves línies d'investigació pel seu
diagnòstic i tractament.
INICI
