 DISTRIBUCIÓ DELS SNPs AL GENOMA DE Mus musculus
DISTRIBUCIÓ DELS SNPs AL GENOMA DE Mus musculus
Els SNPs (Single Nucleotids Polimorphisms), com ja s'ha dit anteriorment, es corresponen amb el canvi d'un únic nucleòtid dintre de la seqüència genòmica. Per poder referir-nos al concepte polimorfisme, aquests canvis han de tenir lloc com a mínim en l'1% de la població. Així doncs, per exemple, una A podria ser substituïda per una C, una T o una G, i això suposaria un augment de la variabilitat.
El nostre propòsit és estudiar quina distribució presenten aquests canvis al llarg del genoma de Mus musculus.
Ens podem preguntar "per què resulta interessant descriure i avaluar la localització dels SNPs a aquesta espècie en concret?"
La resposta la trobem quan analitzem les implicacions que suposa la variabilitat creada pels SNPs. En el camp de la recerca s'estan investigant moltes malalties humanes relacionades amb els SNPs, i és important disposar de models animals per l'experimentació. Donat que existeix una elevada homologia entre el genoma d'Homo sapiens i el de ratolí, resulta de molt interés la ubicació dels SNPs en Mus musculus, ja que aquest podria ser un bon candidat experimental.
El 5 de Desembre del 2.002 es va publicar més del 95% de la seqüència de Mus musculus a la revista Nature. S'han trobat al voltant d'uns 30.000 gens, dels quals només uns 300 són específics de l'espècie. El material genètic es distribueix en 19 autosomes i 2 cromosomes sexuals, però com la seqüenciació es va portar a terme a partir d'un ratolí femella, no es disposa de cromosa Y.
Amb aquest estudi, hem volgut trobar resposta a una sèrie d'interrogants:
- On es concentren la majoria dels SNPs anotats al genoma de ratolí? Presenten més variabilitat les regions no codificants que les codificants? Té això alguna repercussió des del punt de vista biològic?
- Són tots els cromosomes igual de variables?
- Segueixen els cromosomes una distribució normal pel que fa a la quantitat d'SNPs?
- I què podem dir dels gens, segueixen una distribució normal respecte a la quantitat d'SNPs?
- Existeixen gens que destaquen per l'elevada freqüència d'SNPs? Què pot dir-nos això sobre la seva funció?
- Fitxer refGene.txt : Conté una llista de 8.555 gens coneguts i anotats amb el següent format:
NM_geneid | cromosoma | reverse/forward | inici del transcrit | final del transcrit | inici
CDS | final CDS | número d'exons | inici de tots els exons ordenats i separats per comes |
finals dels exons
- Fitxer snpNih.txt : Conté un llistat de 100.823 SNPs enregistrats i amb el següent format:
Referència | cromosoma | iniciSNP | finalSNP | identificador
(*) En una revisió més acurada d'aquests dos arxius hem pogut apreciar la presència d'identificadors repetits tant en el cas dels gens com en el cas dels SNPs. Això té repercusions sobre el resultats finals.
(**)També cal destacar la presència d'una sèrie de gens i SNPs que no s'han pogut ubicar en cap cromosoma, el que ha fet que s'hagin agrupat dins d'un cromosoma designat com chrUn.
La informació extreta d'aquests dos fitxers ens proporciona les eines necessàries per l'estudi dels SNPs en Mus musculus. La idea principal és poder comparar aquests dos fitxers amb la finalitat d'ubicar els SNPs al llarg del genoma de ratolí. En primera instància, cal tenir en compte que tant els gens com els SNPs tenen una posició definida dintre el cromosoma al qual pertanyen, i aquest serà el punt de partida per establir una relació entre amdós arxius.
De les dades contingudes en el fitxer dels SNPs tan sols en farem ús d'aquelles que es corresponen amb:
- Cromosoma (columna 2).
- Posició dels SNPs dintre del cromosoma (columna 4). Els SNPs es corresponen amb una substitució nucleotídica singular, però per convenció s'han de registrar amb una posició inicial i una final. Donat això, nosaltres treballarem amb la quarta columna que és la realment informativa.
- Identificador (columna 5).
Pel que fa al fitxer dels gens utilitzem totes les dades disponibles. Per una banda, els límits dels gens els establim amb les posicions cromosòmiques d'inici i final del transcrit, i per l'altra, definim la regió codificant com l'interval que comprèn el CDS. Entre transcrit i CDS trobaríem els UTRs, els quals no codifiquen per proteïna però, estan presents al mRNA i desenvolupen un paper important en la seva traducció, afavorint el reconeixement d'aquest pel ribosoma. El CDS comprèn les posicions dels introns i dels exons, ja que ens estem referint a posicions cromosòmiques, però tot i així, cal tenir en compte que només seran els exons els que donaran lloc a la seqüència aminoacídica de la proteïna, després de tenir lloc un procés d'splicing. Moltes vegades els exons s'extenen més enllà dels límits del CDS i nosaltres al tractar les dades els considerarem com a UTRs.
Per poder determinar quina és la distribució dels SNPs al llarg del genoma de Mus musculus i establir vincles entre els dos fitxers de partida, elaborem un programa en llenguatge Perl. La potència d'aquest llenguatge de programació es basa en la possibilitat d'establir estructures de dades complexes (com ara la utilitzada en el nostre programa), i en la utilització d'expressions regulars. No ens ha calgut ordenar els fitxers inicials ja que l'estructura que segueix el programa està basada principalment en la utilització de hashes (són anàlegs als vectors però indexen els valors per paraula i no per posició). Aquest fet ens permet un anàlisi molt més ràpid i eficient de les dades sense necessitat de tractar-les previament. La versió complerta del programa s'adjunta en el següent enllaç: El_cerca_SNPs.pl
Seguidament s'analitzaran cadascun dels passos seguits en l'elaboració del programa.
1. Vector predefinit ARGV:
- Mitjançant la utilització del vector ARGV introduïm els 2 fitxers, amb els quals haurà de treballar el programa en el moment de cridar-lo.
- Això permetrà la utilització del programa amb fitxers que presentin el mateix format però amb informació diferent. El fitxer que ocupa la posició 0 del vector predefinit ARGV es correspon amb el que conté la llista dels gens. El segon fitxer que ocupa la posició 1 del vector es correspondria amb els SNPs.
2. Creació i definició del hash %REFGENE:
- Obrim el fitxer.
- La informació està organitzada en diferents camps que es corresponen amb les columnes del fitxer. Elaborem un vector que contingui tota aquesta informació i, a partir d'aquest construïm un hash amb el nom de %REFGENE en el qual tenim com a paraula clau el cromosoma ({$chr}).
- Abans de la creació del hash ens assegurem de què aquest no hagi estat definit prèviament.
- Aquest hash creat, apunta a un altre hash que té com a paraula clau l'identificador del gen ({$gid}).
- Aquest segon hash presenta associats com a valors singulars les diferents posicions que ocupen en el gen les regions codificants o no codificants.
- Reassignem aquestes posicions a diferents variables.
3. Creació i definició del hash %SNPNIH(&obrir_fitxer_SNPs &ordenar_array_SNPs):
- Obrim el fitxer.
- La informació la trobem també organitzada en diferents camps (columnes del fitxer). La introduïm en un vector i, a partir d'aquest construïm un hash amb el nom de %SNPNIH. La paraula clau d'aquest segon hash és també el cromosoma ({$chr}) on es localitza l'SNP.
- Abans de l'elaboració del hash ens assegurem que aquest no hagi estat definit prèviament.
- Aquest hash creat, apunta a un array que conté la següent informació del fitxer dels SNPs: l'identificador ($id) i la posició que ocupa el polimorfisme dintre del cromosoma ($posició).
- El següent pas és ordenar aquest array, de manera que vagi d'aquells SNPs que tenen una posició més baixa dintre el cromosoma a aquells que ocupen una posició més alta. Així el programa és més eficient, ja que quan volguem trobar els SNPs que cauen dintre un determinat gen, no haurem de revisar tota la llista. Començaríem a mirar pel principi de l'array dels SNPs i quan arribèssim a un polimorfisme que té una posició superior a la del final del transcrit del gen, pararíem de buscar.
4. Tractament de dades(&tractament_dades):
Extraiem la informació continguda en els hashes que hem construït amb anterioritat.
- Assignem a una variable totes les paraules claus del hash %REFGENE. Aquesta variable funciona com un nexe d'unió entre els dos hashes, ja que %SNPNIH també té com a paraula clau la ubicació cromosòmica dels SNPs.
- Elaborem una composició que va fent iteracions (utilitzant el foreach) i mirant per cadascun dels cromosomes, els gens, i per cadascun d'aquests analitza tots els possibles SNPs classificant-los en les diferents regions gèniques que estem estudiant.
- A mesura que obtenim la distribució dels SNPs en els diferents gens introduïm aquesta informació en un hash %RESULTAT. Aquest hash té com a paraula clau el cromosoma on estaria el gen i apunta a un altre hash amb l'identificador del gen com a paraula clau. A la vegada el hash secundari apunta a un array que conté el nombre d'SNPs presents en cadascuna de les regions estudiades del gen en qüestió.
5. Imprimint resultats(&impimir_resultats):
En aquest pas tan sols ens cal extreure la informació que s'obté durant el tractament de dades i que hem organitzat en el hash %RESULTAT.
Tal i com s'ha indicat amb anterioritat la nostra finalitat és analitzar la distribució dels SNPs en el genoma de Mus musculus. Per això ens ha calgut la creació del programa distribucio_SNPs.pl, prèviament explicat.
En total, dels 8.555 gens del fitxer refGene.txt tenim constància de 8.472 gens al fitxer dels resultats, donat que hi ha identificadors que es troben repetits al fitxer original. En aquests resultats tan sols es mostren un total de 19.222 SNPs dels 100.823 continguts al fitxer original (snpNih.txt), és a dir, tan sols hem trobat la localització d'aproximadament un 10% dels SNPs. Aquest fet és degut a què:
Per tal de poder fer un anàlisi estadístic d'aquests resultats ens ha calgut la realització de dos petits programes addicionals al programa principal.
Però previ a tot això, ha estat necessari un tractament de les dades amb Shell i les comandes utilitzades s'especifiquen a continuació.
Ara tractarem d'explicar pas a pas, de manera resumida, quin és el funcionament del programa estadistica.pl:
1. Utilització del vector ARGV:
2. Primera funció: Fa la suma del total dels SNPs.
3. Segona funció: A partir del valor que hem obtingut de la funció anterior, ens calcularà la proporció dels SNPs respecte el total que s'han descrit per tot el genoma de Mus musculus.
Fem un altre programa, ara anomenat programa_dades_pergens.pl que utilitza, aprofitant tots els coneixements que hem adquirit amb el programa principal, una estructura de dades basada en un hash. S'ha de tenir en compte que això es podria fer com una continuació del programa distribucio_SNPs.pl, però hem considerat més oportú que els programes per l'anàlisi de les dades haurien d'anar per separat. El programa s'encarrega de fer una suma del total d'SNPs que trobem a cada gen. És a dir, a l'arxiu dels resultats tenim la quantitat d'SNPs que tenen els gens, però separat per les regions gèniques estudiades. Ara es tracta de fer una suma del total per cada gen.
Els passos que segueix el programa són els següents:
1. Vector predefinti ARGV:
Introduïm pel teclat el fitxer dels resultats.
2. Construir hash %ESTADISTICA:
Elaborem aquest hash amb la informació continguda al fitxer dels resultats. Ja s'ha comentat l'estructura que presenta aquest fitxer anteriorment.
3. Tractament de dades:
Cridem per cadascun dels identificadors els valors singulars associats a la paraula clau del hash, i fem una suma de tots aquests, que assignem a una nova variable.
El resultat obtingut de l'aplicació d'aquest programa es mostra al fitxer: resultatSNPs1.txt i la seva estructura consisteix en diferents línies. Cadascuna d'aquestes conté les dades per un únic gen. El primer que trobem a cada línia és l'identificador del gen, per exemple NM_011605, seguit del cromosoma al qual pertany, juntament amb l'
Concretament el programa estadistica.pl , ens facilita la distribució dels SNPs de dues maneres diferents:
D'una banda, tenim les comandes que ens permeten a partir del fitxer dels resultats, extreure tan sols la informació numèrica pel cas del càlcul de les proporcions d'SNPs en el genoma sencer. Aquestes són les següents:
Per una altra banda, tenim la comanda utilitzada per fer els càlculs numèrics que ens permeten fer un anàlisi per cromosoma dels nostres resultats; i és la que segueix: bash-2.05$ cut -f 2 resultatSNP1.txt > UTRs.txt
bash-2.05$ cut -f 3 resultatSNP1.txt > exons.txt
bash-2.05$ cut -f 4 resultatSNP1.txt > introns.txt
bash-2.05$ cut -f 5 resultatSNP1.txt > donors.txt
bash-2.05$ cut -f 6 resultatSNP1.txt > acceptors.txt
egrep '(chr_\b)' resultatSNP1.txt > chr_resultatSNP1.txt
NOTA: Aquesta comanda ens permet extreure per separat els resultats dels gens que estan en els diferents cromosomes del genoma de Mus musculus. La part d'aquesta comanda especificada com chr_ s'hauria de canviar per cadascun dels cromosomes per exemple: chr1, chr2... S'obtenen així 21 fitxers. Amb aquests fitxers es repeteix el procediment utilitzat anteriorment per extreure els valors numèrics i fer la suma i la proporció del total d'UTRs, exons, introns, donors i acceptors per cadascun dels cromosomes.
Abans d'entrar en detall en el que seria realment l'anàlisi dels resultats, volem destacar un aspecte concret. Tal i com ja s'ha comentat, hi ha tant gens com SNPs que no s'han pogut ubicar a cap cromosma i s'han agrupat en el cromosoma unrandom (chrUn). Aquests gens i SNPs provenen de la seqüenciació de segments genòmics, dels que es desconeix la procedència. Per aquest motiu, les seves posicions no són significatives.
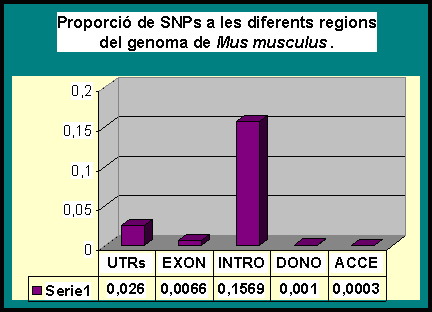
Gràfic 1.1:
En aquest gràfic es mostra quines són les regions genòmiques més variables. Podem veure com la zona on es concentren la gran majoria dels polimorfismes estudiats són les regions intròniques i els UTRs. Totes dues regions es caracteritzen per no formar part de la proteïna final, és a dir, no tenen tanta pressió de la selecció natural, i per tant són més susceptibles al canvi.
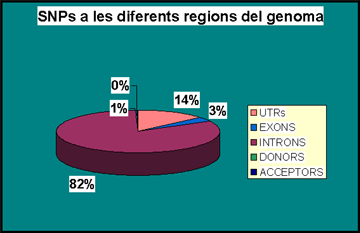
Gràfic 1.2:
Aquest gràfic és complementari a l'anterior. En aquest cas, hem volgut representar les proporcions d'SNPs per les diferents regions estudiades, però sense tenir en compte tots els SNPs trobats al genoma de ratolí, és a dir, hem obviat aquells SNPs que cauen en regions intergèniques. Tal i com s'aprecia, la gran majoria dels SNPs estan ubicats en els introns, corresponent-se amb un valor del 82%. Tan sols tenim un 14% d'SNPs en UTRs i un 3% per les regions codificants, mentre que la presència d'SNPs a les regions d'splicing és pràcticament nul.la. Tot i que aquí el percentatge de polimorfismes per les regions d'acceptors és del 0%, ja sabem pel gràfic anterior que existeix una certa representació d'SNPs en aquestes zones.
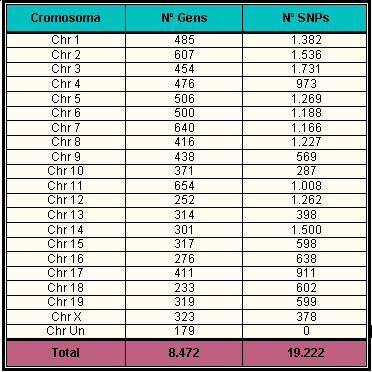
Taula 1
En la següent taula es mostren el nombre de gens que hi ha en cada cromosoma del genoma de Mus musculus i el nombre d'SNPs per cadascun d'aquests cromosomes.
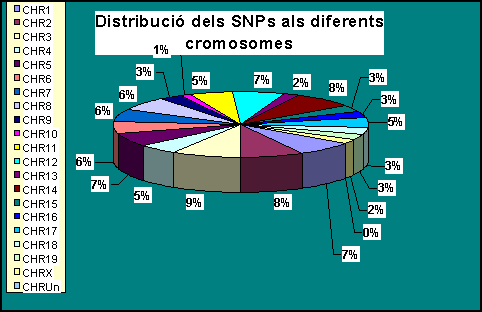
Gràfic 2.1:
Aquest segon gràfic ens mostra la variabilitat en el genoma de Mus musculus. La distribució d'SNPs ens la mostra per cadascun dels cromosomes, en forma de tant per cent (%).
S'observa que el cromosoma més variable és el cromosoma 3 . Aquest presenta 454 gens amb un total de 1731 SNPs repartits en les regions gèniques estudiades (Gràfic 2.2). Representa el 9% del total d'SNPs del genoma. Per contrapartida, el cromosoma menys variable és el cromosoma 10, amb 371 gens i 287 SNPs; aquest només conté un 1% del total d'SNPs.
Però tot i així, veiem com es dóna una distribució més o menys homogènia en els cromosomes (aquesta idea es veu més clara al Gràfic 3). No tenim cap cromosoma que destaqui per la seva elevada variabilitat, de la mateixa manera que tampoc existeix cap que s'hagi mantingut més conservat al llarg de l'evolució. Però, òbviament, cada cromosoma no presenta el mateix nombre de gens, el que serà indicatiu de l'existència de gens que tindran un nombre d'SNPs més elevat que d'altres. Tot i això, veiem com la selecció ha actuat de manera molt similar a tot el genoma.
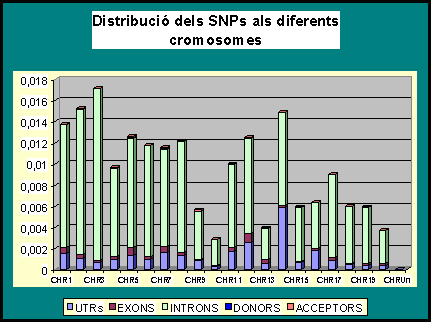
Gràfic 2.2:
Aquest és un gràfic que ens complemeta la informació que ens ha proporcionat l'anterior. L'alçada de les barres reflexa la quantitat d'SNPs per cada cromosoma (pel que fa als números d'aquest eix vertical s'ha de tenir en compte que són proporcionals al total d'SNPs revisats a cada cromosoma). A més també, es mostra on es troben concentrats la major part dels polimorfismes dintre de cada cromosoma, i com ja mostrava el gràfic 1, tenim que la major part es troben a les regions intròniques i UTRs, destacant, sobretot, la presència en introns.
Ens crida l'atenció el cromosma 14, que presenta una proporció molt més elevada d'SNPs a les regions UTR, que no pas la resta de cromosomes.
Cal remarcar, també la presència pràcticament nul.la d'SNPs als llocs d'splicing (donors i acceptors). Per exemple en els cromosmes 3, 9, 10, 11, 14, 16 i en el cromosoma X no cauen SNPs en les regions donadores d'splicing.
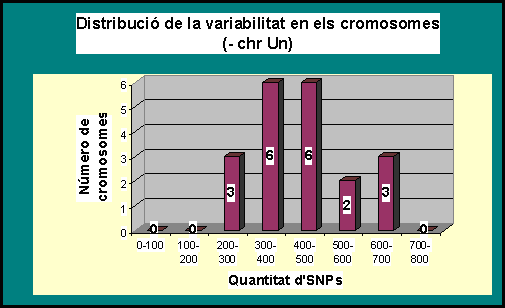
Gràfic 3:
Passem ara a mirar quina distribució segueixen els SNPs al llarg dels cromosomes. S'observa una distribució semblant a la "normalitat" (campana de Gauss). Veiem que 12 cromosomes d'un total de 20 (no tindrem en compte el chrUn, ja que com s'ha comentat no és significatiu) tenen entre 300 i 500 SNPs. Ens queden doncs, 8 cromosomes que presenten un nombre de polimorfismes superior a 500 o inferior a 300. Com es pot veure la distribució es troba desplaçada una mica cap a valors màxims, és a dir, tindrem més cromosomes que presenten una quantitat de SNPs superior a 500 que no pas amb quantitats menors de 300. A més, no hi ha cap cromosoma que no presenti cap SNP.
Taula 2
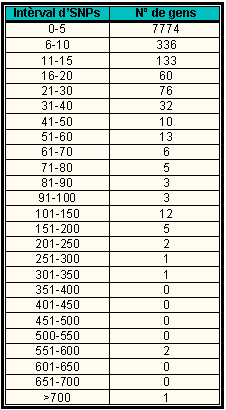
En aquesta altra taula es mostren els valors amb els quals hem treballat posteriorment per fer un estudi de la distribució dels SNPs al llarg dels diferents gens del genoma de ratolí
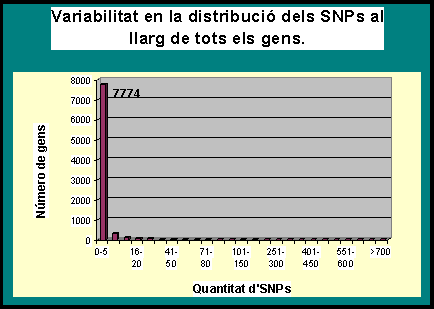
Gràfic 4.1:
Mirem ara quina és la distribució dels polimorfismes al llarg dels diferents gens. Aquesta distribució no segueix una distribució normal, sinó tot el contrari. Tenim la majoria dels gens concentrats en un únic interval que va de 0 a 5 SNPs per gen, això és indicatiu de la baixa variabilitat que presenten les regions gèniques.
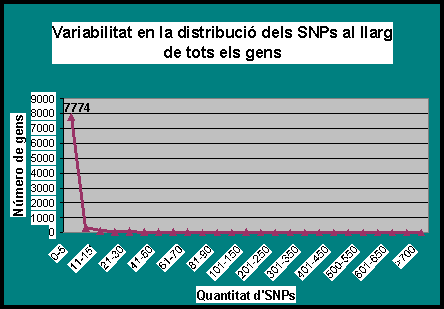
Gràfic 4.2:
Aquest gràfic ens mostra el mateix que l'anterior, però ho hem volgut destacar d'altra manera amb la finalitat de fer més visual la distribució que segueixen els gens. Com s'observa, a diferència del que obteníem amb els cromosomes, en aquests cas els gens no segueixen una distribució normal.
Ens ha cridat molt l'atenció la presència d'un gen que destaca per l'elevada quantitat d'SNPS que presenta respecte la resta de gens del genoma de Mus musculus. Aquest gen es troba al cromosma 12, i el seu transcrit engloba la regió ubicada entre les posicions 29623298-29659762. L'identificador d'aquest gen és NM_013464, el que ens ha permès fer la cerca a Entrez per saber a quina proteïna es correspon. Hem trobat que es tracta de l'Aryl Hydrocarbon Receptor_Interacting Protein Like 1(AIPL1). D'aquesta proteïna encara no es coneix ben bé la seva funció, però, la presència del domini TRP indica que pot estar implicada en el transport nuclear o bé, podria tractar-se d'una acitvitat tipus chaperona que participa en el plegament de proteïnes.
En total conté 830 SNPs repartits en: 179 en UTRs, 82 en exons, 559 en introns, 3 en donors i 7 en acceptors. Aquests valors obtinguts, ens fan pensar que potser, aquesta proteïna donada la funció que desenvolupa pugui acceptar més variabilitat.
La variabilitat també és elevada als UTRs, encara que en menor grau que als introns. Tal i com hem pogut demostrar en els Gràfics 1.1 i 1.2, existeix una predominància molt important d'SNPs a introns; estem parlant d'un 82% enfront de només un 14 % per les regions d'UTRs. Aquesta diferència encara és més abismal quan ho comparem amb la resta de regions. Els UTRs tampoc formen part del producte proteic, momés tenen participació en la unió de la maquinària de traducció cel.lular, per l'expressió de les proteïnes. Això explica que els nucleòtids que els conformen estiguin una mica més conservats que els introns, tot i no arribant al grau de conservació de les regions exòniques.
Després de l'estudi de la distribució dels SNPs al llarg del genoma volem constatar el fet de què les regions exòniques, és a dir, les que codifiquen per proteïna, són de les més conservades. Les que realment s'han mantingut al llarg de l'evolució amb menys variacions han estat les regions implicades en l'splicing. Amb aquest fet hem pensat que podien ocòrrer dues coses diferents. Una d'elles és que trobem menys nucleòtids que pertanyin a regions d'splicing que nucleòtids que es troben en exons, i això pot fer reduir la proporció de polimorfismes en aquestes zones del genoma. Per una altra banda, és possible que un canvi d'un nucleòtid dintre de la part codificant sigui més suportable, mentre que pel contrari, un sol canvi en les regions d'splicing pot donar lloc a un proteïna totalment diferent o truncada.
Cal tenir en compte que per aquest estudi no s'han avaluat directament les regions intergèniques. Com només tenim un 10% d'SNPs ubicats a les regions estudiades, la resta que serien la majoria, estarien inclosos dintre aquestes zones que es troben fóra dels gens. Amb la qual cosa, es torna a fer palès que les regions més variables són les no codificants.
Des del punt de vista biològic aquest resultat és significatiu ja que la selecció natural recolza la conservació d'aquelles parts del genoma codificants o amb un paper funcional important.
L'anàlisi de la variabilitat estudiada per cada cromosoma ens nostra que les diferències entre ells no són apreciables. Tenim que la majoria dels cromosomes en Mus musculus tenen entre 300 i 500 SNPs. Per tant, la quantitat d'SNPs és molt similar a tots els cromosomes, no hi ha cap que destaqui significativament per sobre els altres. A més, continuem veient com es manté la baixa variabilitat a les regions que donen lloc als exons i als donors i acceptors dels llocs d'splicing.
Pel que fa a la distribució de la quantitat de polimorfismes no per cromosomes sinó per gens veiem que la majoria, és a dir, un 99% dels que han estat ubicats del genoma de ratolí, presenten una quantitat d'SNPs baixa (0-50 polimorfismes). Després tota la resta dels gens presenten diferents quantitats fins un valor màxim de 830 SNPs. Aquest seria el gen més variable del genoma de ratolí i ja ha estat comentat prèviament. Les diferències en la distribució dels SNPs en cromosomes i gens podrien ser conseqüència de què, encara que la majoria de gens presenten la mateixa quantitat de SNPs (0-50), hi ha cromosomes amb una major quantitat de gens.
Com a conclusió final dir que els SNPs suposen una font de variació dins la població, i aquesta variabilitat és més acceptada a les regions no codificants.


Per qualsevol cosa no dubteu en enviar-nos un mail:
cristina.carreno01@campus.upf.es
blanca.reyes01@campus.upf.es