 DNA Microarrays i Càncer de Pròstata
DNA Microarrays i Càncer de Pròstata
DNA Microarrays i Càncer de Pròstata Per cortesia de Amarant Martínez Carrió i Jordi Martínez-Quintanilla Martínez. 4t Curs Biologia Humana. Curs 2002-2003.
El càncer de pròstata és el tipus de càncer més extès i dels més greus en l'home. Malgrat existeixen proves, com el screening del PSA, que identifiquen la malaltia, aquestes no són prou específiques. La caracterització dels perfils d’expressió gènica mitjançant la hibridació de microarrays pot permetre distingir molecularment en neoplàsies prostàtiques els gens involucrats en la carcinogènesi de pròstata (potencials gens diana de disseny de fàrmacs), elucidar biomarcadors clínics, i porta necessàriament cap a una millora de la classificació dels càncers de pròstata.
Recentment, dos grups diferents han utilitzat els microarrays de DNA per tal d'identificar un conjunt de gens predictius del desenvolupament de metàstasis a curt termini. En concret, van estudiar l'expressió de més de 15000 gens en grups de teixits de pròstata que incluien mostres metastàsiques i mostres no malignes. Tant un grup com l'altre proposen gens que poden ésser utilitzats com a marcadors tumorals. L'objectiu del projecte és revelar un conjunt de gens comuns als dos grups que podrien ser bons marcadors tumorals.
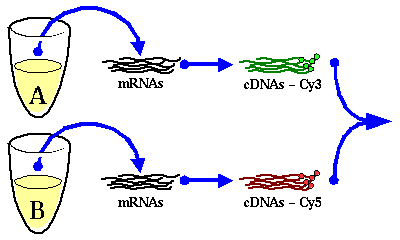
Els microarrays són una col·lecció de biomolècules ordenades ortogonalment sobre un suport sòlid miniaturitzat, és a dir, els microarrays són una peça petita que conté molts pous a cada un dels quals s´ha introduit una seqüència de cDNA de cadena simple d´un gen determinat.
Els microarrays és una de les tècniques més recents amb les que compta la biologia molecular. Va ser desenvolupada a finals dels anys 80 i és una eina molt útil per trobar diferències d´expressió gènica entre cèl.lules o teixits que estan sotmesos a diferents condicions. Així podem trobar els gens que estan sobreexpresat o reprimits en les diferents fases del cicle cel·lular, durant el procés de desenvolupament dels òrgans, en resposta a diferents estímuls externs (temperatura) o en diferents condicions patològiques, com per exemple el càncer. A partir d´aquests patrons d´expressió es poden predir certes condicions patològiques.
 |
 |
 |
Hi ha diversos tipus d´agrupament o clusterings utlitzats en microarrays:
El clustering és un procés d´agrupament de dades de tal forma que els objectes d´un cluster tinguin una similaritat alta entre ells i baixa entre els objectes d´altres clustering.
És un agrupament aglomeratiu que genera un arbre jeràrquic. És un sistema molt simple i els seus resultats són molt fàcils de representar mitjançant programens com el Tree View.
És un clustering divisiu en el que es parteix d´un nombre predeterminat de grups de gens o condicions (igual que en els altres programas de Cluster) i calcula un vector d´expressió promig que es va refinant per càlcul reiteratiu. No produeix arbre, només t´agrupa els gens segons la seva expressió diferencial.
És un clustering divisiu basat en xarxes neuronals. És un mètode que optimitza la separació de grups en base a geometria predefinida sobre la qual s´entrenen vectors de referencia. Maximitza la convergència entre vectors i punts de cada cluster.
El nostre treball ha estat basat en l'estudi de dos articles. Aquests articles són els següents:
El grup de recerca dirigit per Dhanasekaran va examinar mitjançant l’ús de microarrays de cDNA, els perfils d’expressió gènica de més de 50 mostres de teixit prostàtic normal i neoplàsic, en les que es va analitzar l’expressió d’un total de 9.984 gens. Les mostres analitzades foren les següents: 4 mostres de BPH (Glàndules Hipertrofiades Benignes), 8 mostres de NAP (Teixit Prostàtic Normal Adjacent), 1 pool comercial de teixit normal de pròstata de 19 individus, 1 mostra de prostatitis, 11 PCA (mostres de càncer de pròstata localitzat), 7 MET (mostres de càncer de pròstata metastàsics), 3 línies de càncer de pròstata metastàsic : DU-145, LnCAP, PC3. A més, es van incloure en l’estudi 28 mostres de teixit de pròstata addicional. Com a mostres de referència es van utilitzar 2 pools diferents: 1 pool de NAP de pacients amb càncer de pròstata i un pool comercial.
Un cop obtinguts els valors d’expressió per cada gen, es van utilitzar diferents mètodes estadístics (t-test) per centrar l’anàlisi només en els gens que tenen una expressió significativament major en les mostres tumorals. Per agrupar els gens i les mostres experimentals segons les relacions d’expressió gènica obtingudes al microarray es va utilitzar un algoritme de clustering jeràrquic. Per visualitzar aquestes dades van fer servir un dendrograma, on el patró de les branques reflectia el grau de relació dels gens.
El resultat de l’estudi va ser que hi havia un conjunt de gens sobreexpressats que podien servir com a marcadors de càncer de pròstata. Aquest set de gens és el següent: HPN (hepsina), LIM (ENIGMA), Pim1 (proto-oncogen), MYC (proto-oncogen), Sintasa d’ŕcids grassos, TIMP2, HEVIN, RIG, THBS1 (thrombospondin-1), MTA-1 (metastasis-associated1), MYBL2 i FLS353. Aquests gens identificats per anàlisi de microarray van ésser corroborats posteriorment mitjançant Northern Bloot.
El grup de recerca dirigit per Welsh va caracteritzar el càncer de pròstata primari mitjançant la monitorització dels nivells d’expressió de més de 8900 gens en teixits normals i teixits malignes. La finalitat del grup d’investigadors era augmentar l’especificitat per detectar el càncer de pròstata invasiu i trobar noves dianes terapèutiques específiques del càncer de pròstata. Les mostres que van utilitzar van ser les següents: 25 mostres derivades de teixit de càncer de pròstata, 9 mostres provinents de teixits prostàtics no malignes, i 21 línies cel·lulars. Com a control van utilitzar les següents línies cel·lulars: PrEC (Epiteli Prostàtic Normal), hPr1 (cél·lules infectades amb papiloma), CAF1598, 1303, 1852, 2585 (fibroblasts adjacents a l’adenocarcinoma), BPHF 1598 (Glàndules Hipertrofiades Benignes), PrSC (Cèl·lules de l’Estroma Prostàtic).
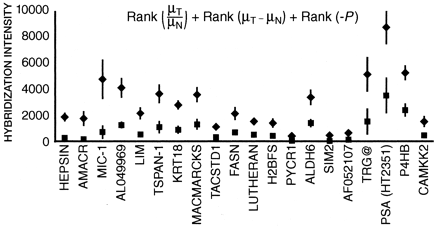
Els valors d’expressió obtinguts (intensitat d’hibridació) van ser normalizats per cada gen. Gens i mostres van ésser agrupades mitjançant clustering jerŕrquic. L’expressió diferencial dels gens en teixits benignes i malignes de pròstata va ésser estimada mitjançant un algorsime basat en les diferències d’intensitat de la hibridació (utumor - unormal), el quocient d’intensitats d’hibridació (utumor / unormal) i el resultat d’un t-test de dades no aparellades dels nivells d’expressió en tumor i teixits normals. Els gens van ser puntuats segons aquests tres parŕmetres i ordenats segons la suma de les tres puntuacions. Amb els valors obtinguts es va dibuixar un dendrograma en el que la similaritat total és proporcional a la longitud de les branques verticals.
El perfils d’expressió gènica obtinguts van revelar l’existència d’un grup d’uns 400 gens que es troben sobreexpressats en tumors. Els més importants d’aquests gens estan representats en la figura següent:
Els passos que hem dut a terme per realitzar el nostre treball estan resumits als següents punts:
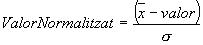 |
Els resultats que hem obtingut d'estudiar l'expressió genètica diferencial exposada en ambdós estudis és que hi ha 5 gens compartits que es sobreexpressen en teixits tumorals. Aquests 5 gens són l'Aminocylasa 1 (ACY1), el Factor d'Elongació 1 gamma (EEF1G), l'Hepsina (HPN), una proteïna similar a la Proteïna Quinasa C (LIM) i una proteïna de la família de les Serin-Treonin Quinases (CAMKK2). L'expressió específica de cada un d'aquests gens als diferents teixits és la següent
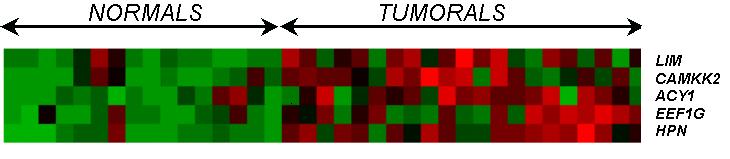 |
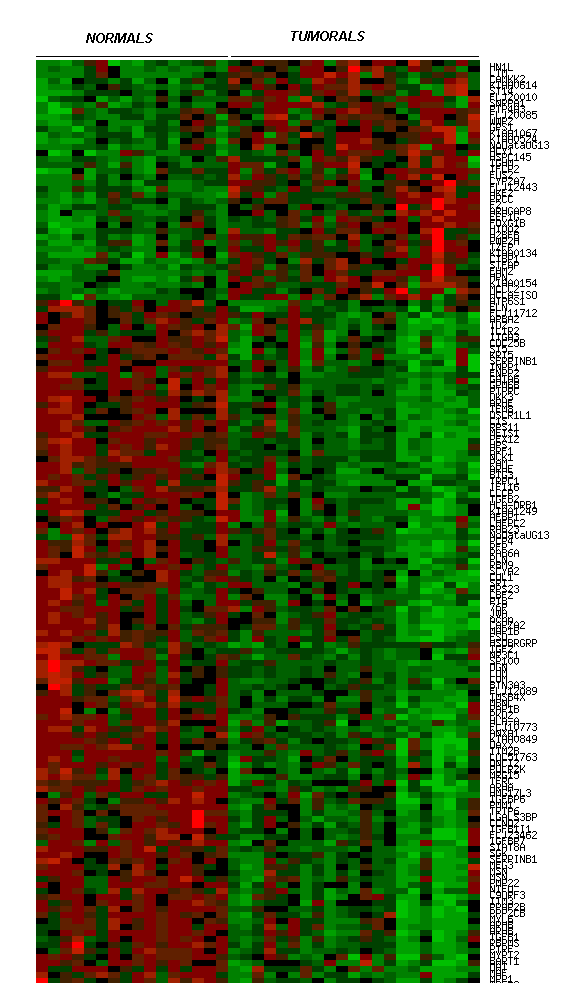
Fig. 5 Expressió diferencial dels 5 gens compartits als teixits utilitzats en l'anàlisi de Dhanasekaran.
 |
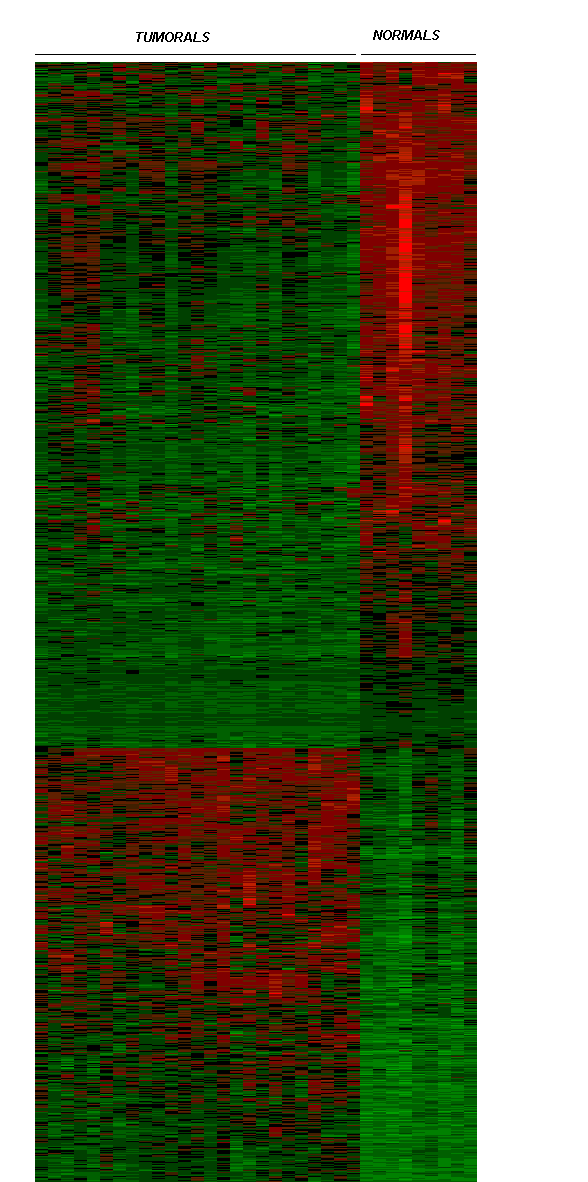
Fig. 6 Expressió diferencial dels 5 gens compartits als teixits utilitzats en l'anàlisi de Welsh.
A partir del codi GenBank d´aquests gens anem al programa GO que es un programa que ens representa un gràfic de les funcions metabòliques en les que estan implicades els gens que li entrem. El gràfic que obtenim dels 5 gens seleccionats és el següent:
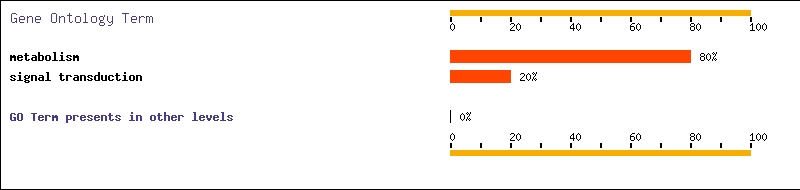
| Traducció de senyals (GO:0007165) | LIM |
| Metabolisme (GO:0008152) | ACY1, CAMKK2, EEF1G, HPN |
Veiem que 4 dels 5 gens estan implicats en metabolisme i només un gen està implicat en transducció de senyals. Cap dels 5 gens està implicat en cicle cel·lular, ni en proliferació cel·lular, ni en oncogènesi que són funcions, en principi, més relacionades amb càncer.
També busquem informació sobre els gens a GenBank i sobre les proteïnes a Swissprot.La informació que obtenim és la següent:
Aquest gen codifica per l´aminoacylasa 1 d´Homo sapiens. L´aminoacylasa-1 és un enzim citosòlic homodimèric que s´uneix a zenc i està involucrat en el metabolisme dels aminoàcids i en la degradació de proteïnes. Concretament, catalitza la hidròlisis de l´aminoàcid L-acilat a aminoàcid L i un grup acil. ACY1 ha estat assignat al cromosoma 3p21.1, una regió reduïda a homozigositat en càncer de pulmó de cèl·lules petites (SCLC), i s´ha vist que la seva expressió es troba disminuida en les línies cel·lulars d´aquest càncer.
Aquest gen codifica per una subunitat del complexe 1 del factor d´elongació gamma, que és el responsable d´apropar els tRNAs al ribosoma. Aquesta subunitat conté un domini N-terminal de la glutatión transferasa, que pot estar implicat en la regulació de l´ensamblatge de complexes amb multisubunitats que contenen aquest factor d´elongació.
Hepsin és una serin proteasa transmembrana d'Homo sapiens.
LIM és una proteïna similar a la proteïna kinasa C que s´uneix a Enigma.
El producte d´aquest gen pertany a la família de les Serin-threonin kinases. Aquesta prote&imul;na juga un rol en la cascada de quinases depenents de calci/calmodulina (CaM) fosforilant la quinasa downstream CaMK1 i CaMK4. A partir de 7 transcrits es poden codificar 6 isoformes diferents.
Obtenim molt poca informació sobre aquestes proteïnes perquè estan molt poc estudiades. Per aquest motiu pensem fer el mateix gràfic al programa GO amb els sets de gens que havíem obtingut en el punt 5 i 6. Aquests sets són els gens que estaven sobreexpressats en tumors abans de comparar el set de Welsh amb el Danasekaran. Així podem veure quina funció predomina en aquests gens. Els gràfics són els següents:
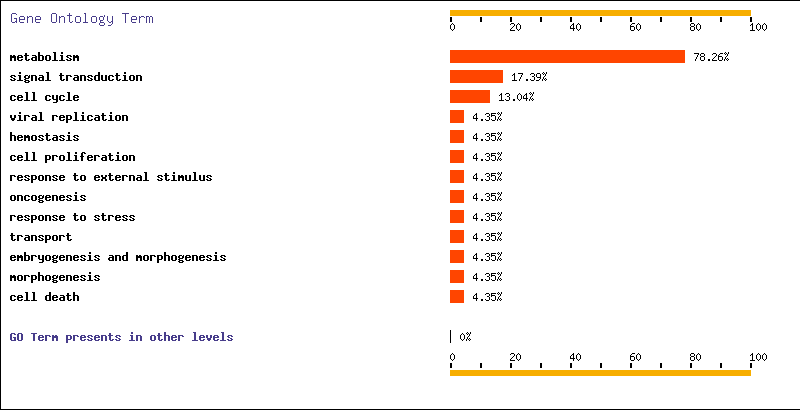
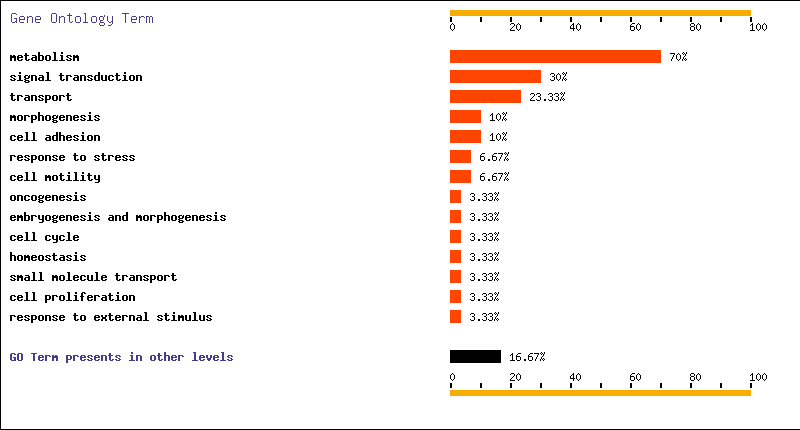
Veiem que als dos sets de gens trobem molts gens que estan implicats en metabolisme (com ACY1,EEF1G,HPN,CAMKK2) i altres que estan expressats en transducció de senyals (com LIM), cicle cel·lular i transport, però amb molta menys proporció. Per aquest motiu pensem que hi ha d´haver vies metabòliques que estiguin implicades en el desenvolupament de càncer de pròstata.
Un altre punt important és comprobar quina p-value tenien inicialment aquests gens en els dos sets de dades. La taula següent ens ho mostra:
| Gen symbol |
|
Codi NM | p-Value Welsh | p-Value Dhanasekaran |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
En aquesta taula veiem una clara diferència entre els valors de p-value de l´experiment de Welsh i els de Dhanasekaran. Això ens indica que el fet d´utilitzar diferents mostres de teixits normals i tumorals pot fer variar els valors resultants d´un experiment de microarrays i, per tant, dos experiments de microarrays fets amb la mateixa finalitat poden donar resultats molt diferents.
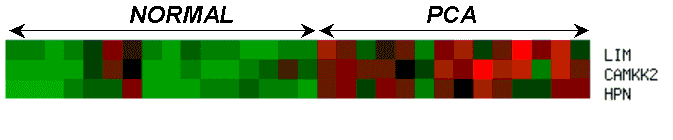
S´ha de tenir en compte que tant els teixits PCA com els teixits metastàsics són tumorals, però es diferencien en que el PCA és un teixit tumoral localitzat i per tant menys maligne que el teixit metastàsic que és molt més greu. Pensem que és interessant intentar obtenir més informació dels 5 gens que hem obtingut comparant les seves expressions entre teixits normals i PCA, entre PCA i teixits metastàsics i entre teixits normals i teixits metastàsics. Per fer això fem un t-test per cada un dels plantejaments (Normal vs PCA, PCA vs Met i Normal vs Met) i eliminem els gens que tenen una p-value superior a 0,001, quedant-nos amb els gens que són significatius.Aquest experiment només el podem fer amb les dades del treball de Danasekaran perquè utilitzen teixits PCA i Metastàsic. En l'experiment de Welsh no es fan diferències entre els estadis dels teixits tumorals que estudien. El fet d´utilitzar les dades d´un únic experiment fa que els resultats obtinguts seguin menys fiables ja que el fet de comparar experiment dóna més credibilitat als resultats. Seguidament mostrem els resultats per cada experiment.
Per fer aquest gràfic hem eliminat els gens ACY1 i EEF1G perquè ens donava un valor de p-value inferior a 0.001. A partir del tres gens que ens queden mirem la seva expressió diferencial i la representem en forma de gràfic. Els resultats són els següents:
 |
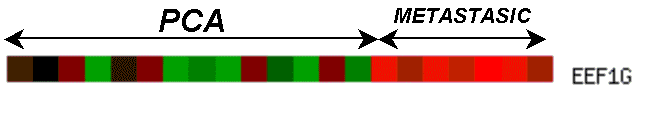
Per fer aquest gràfic hem eliminat tots els gens menys el EEF1G perquè ens donaven un valor de p-value inferior a 0.001. Amb el gen que ens queda mirem la seva expressió diferencial i la representem en forma de gràfic. Els resultats són els següents:
 |
Per fer aquest gràfic hem eliminat els gens ACY1,LIM I CAMKK2 perquè ens donava un valor de p-value inferior a 0.001. A partir del tres gens que ens queden mirem la seva expressió diferencial i la representem en forma de gràfic. Els resultats són els següents:
 |
Les conclusions a les que hem arribat després de realitzar el nostre anàlisi són les següents:

 Una salutació dels qui han creat la pàgina
Una salutació dels qui han creat la pàgina 
{kind=link}
{kind=link}