Summary
- Breast cancer
- Microarray technique
- Summary of the article:
(if you want to link the article click Gene expression profiling predicts clinical outcome of breast cancer) - Our project
introduction
objectives
methods
results
conclusions
- References
- About this document
Breast cancer
National Cancer InstituteAmong women, breast cancer is the most commonly diagnosed cancer after nonmelanoma skin cancer, and the second leading cause of cancer deaths after lung cancer. In 2001, an estimated 192,200 new cases will have been diagnosed and 40,200 deaths from breast cancer will have occurred. A possible genetic contribution to breast cancer risk is indicated by the increased incidence of breast cancer among women with a family history of breast cancer, and by the observation of rare families in which multiple family members are affected with breast cancer, in a pattern compatible with autosomal dominant inheritance of cancer susceptibility.
Formal studies of families (linkage analysis) have subsequently proven the existence of an autosomal dominant form of breast cancer, and have led to the identification of several highly penetrant genes of major effect as the cause of inherited cancer risk in many cancer-prone families. These mutations are rare and are estimated to account for no more than 5% to 10% of breast cancer cases overall. It is likely that other background genetic factors contribute to the etiology of breast cancer. The syndromes associated with an autosomal dominant inheritance of breast cancer risk are hereditary breast and ovarian cancer due to BRCA1 or BRCA2 mutations. Mutations in these genes produce different clinical phenotypes of characteristic malignancies.
Other risk factors for breast cancer include age, previous breast disease, reproductive and menstrual history, estrogen therapy, radiation exposure, diet, and alcohol intake.
In vitro studies of BRCA1 and BRCA2 function suggest a possible role for
these genes in x-ray-induced DNA repair. Mouse cells lacking the BRCA1
protein have been shown to be deficient in repair of oxidative DNA damage
(the kind of damage caused by ionizing radiation), and to have reduced
survival after exposure to x-rays. While human tumor cells deficient in the
BRCA2 protein also demonstrate deficiencies in the repair of
radiation-induced DNA breaks, cells that carry a mutated copy of BRCA2
and a normal copy have normal repair.These preliminary data suggest that
increased sensitivity to radiation could be a cause of cancer susceptibility in
carriers of BRCA1 and BRCA2 mutations. Since mutation carriers are
heterozygotes, however, radiation sensitivity might occur only after a somatic
mutation damaged the normal copy of the gene.
Major Genes
Epidemiologic studies have clearly established the role of family history as
an important risk factor breast cancer.
We now know that some of these cancer families
can be explained by specific mutations in single cancer susceptibility genes.
The recent isolation of several of these genes associated with a significantly
increased risk of breast/ovarian cancer make it possible to identify families
who carry mutations in these genes.
Hereditary breast cancer is characterized by early age at onset (on average
5-15 years earlier than sporadic cases) bilaterality, vertical transmission
through both maternal and paternal lines, and familial association with
tumors of other organs, particularly the ovary and prostate gland.
The search for genes associated with hereditary susceptibility to breast
cancer has been facilitated by the study of large kindreds with multiple
affected individuals, and has led to the identification of several susceptibility
genes, including BRCA1, BRCA2, p53, and PTEN/MMAC1.
BRCA1
In 1990, a susceptible gene for breast cancer was mapped in chromosome 17 (17q12-21) The evidence for the coincident transmission of both breast and ovarian cancer susceptibility in linked families was observed.The BRCA1 gene was subsequently identified by positional cloning methods, and has been found to contain 24 exons that encode a protein of 1,863 amino acids.BRCA2
A second breast cancer susceptible gene, BRCA2, was localized to the long arm of chromosome 13 (13q12.3) through linkage studies.BRCA2 is also a large gene with 27 exons that encode a protein of 3,418 amino acids. Like BRCA1, BRCA2 appears to behave like a tumor suppressor gene with loss of the unmutated allele found in tumor specimens.BRCA1 and BRCA2 Function
Women carrying germline mutations in BRCA1 or BRCA2 have an extremely high lifetime risk of developing breast and/or ovarian cancer. Most of these germline mutations are predicted to produce a truncated protein product. Additionally, in all cancers that have been studied from mutation carriers, the wild-type allele is deleted, strongly suggesting that BRCA1 and BRCA2 are in the class of tumor suppressor genes, i.e. genes whose loss of function can result in neoplastic growth.
Additional evidence that BRCA1 is a tumor suppressor gene is that overexpression of the BRCA1 protein leads to growth suppression in a fashion similar to the tumor suppressor genes p53 and the retinoblastoma gene. While this provides a conceptual framework for understanding the role of mutations in cancer progression, it does little to indicate how these genes normally function to prevent cancer.
Both of these genes code for large proteins. BRCA1 is 1,863 amino acids
producing a protein of about 220kd, while BRCA2 is approximately twice
that size at 3,418 amino acids making a 380kd protein. Both proteins are
normally located in the nucleus and contain phosphorylated residues.
BRCA1 contains only 2 recognizable protein motifs, a RING finger domain
near the N-terminus and a BRCT domain at the C-terminus. RING fingers
are cysteine-rich sequences that coordinate the binding of 2 zinc ions.
This type of domain may facilitate both protein-protein and protein-DNA interactions.
The RING finger in BRCA1 appears to specifically interact with another similar RING
finger protein known as BARD1 that was recently identified based on this
interaction. Both BARD1 and BRCA1 also share another conserved
sequence known as the BRCT domain, a phylogenetically conserved
sequence found in proteins involved in DNA repair and cell cycle
regulation.
Even though BRCA2 is almost twice as long as BRCA1, it has no
recognizable protein motifs and has no apparent relation to BRCA1.
Nonetheless, BRCA1 and BRCA2 appear to share a number of functional
similarities that may suggest why mutations in these genes lead to a specific
hereditary predisposition to breast and ovarian cancer.
Both BRCA1 and BRCA2 are expressed in most tissues and cell types
analyzed, suggesting that it is not the expression pattern that leads to the
tissue restricted phenotype of breast and ovarian cancer. The transcription
of both genes is induced late in the G1 phase of the cell cycle and remains
elevated during the S phase, indicating some role in DNA synthesis.
Both BRCA1 and BRCA2 appear to be involved in the control of meiotic
and mitotic recombination and in the maintenance of genomic stability.
A variety of evidence now points to BRCA1 and BRCA2 being directly
involved in the DNA repair process. Both gene products interact with the
RAD51 protein, a key component in homologous chromosome
recombination and double-strand break repair.Perhaps through this
mutual association with RAD51, BRCA1 and BRCA2 can associate with
each other at sites of DNA synthesis after the induction of DNA damage.
In order to study the function of BRCA1 and BRCA2, homozygous knockout mice have been created for each gene. In most cases, the complete loss of function of either BRCA1 or BRCA2 results in embryonic lethality characterized by a lack of cell proliferation. Cells derived from mouse embryos lacking BRCA1 or BRCA2 are defective in their repair of DNA damage.BRCA2-deficient cells in particular are hypersensitive to radiation and radiomimetics in vitro. BRCA2-defective cell lines were also highly sensitive to chemotherapeutic agents that induce double-strand DNA breaks. These findings are important not only for elucidating the mechanisms of BRCA2 tumor induction, but may also suggest a novel genotype-based approach to choosing therapies for breast cancer in women with BRCA2 mutations.
Finally, both BRCA1 and BRCA2 knockout mice can be partially rescued by crossing with a p53 knockout strain, suggesting that these genes interact with the p53-mediated DNA damage checkpoint. Therefore, the available evidence indicates that both hereditary susceptibility genes are gatekeepers, like p53, which serve to maintain genomic integrity. When this function is lost, it probably allows for the accumulation of other genetic defects that are themselves directly responsible for cancer formation. BRCA1 and BRCA2 can be thought of as true cancer susceptibility genes.
Additional studies have attempted to attribute specific biochemical functions to the BRCA1 and BRCA2 gene products. Both proteins contain regions that are capable of inducing transcription.One of the targets of BRCA1 transcriptional activation appears to be the p21 cyclin-dependent kinase inhibitor, itself a potent suppressor of growth at the G1/S checkpoint.The BRCA2 protein has histone acetyltransferase activity, potentially supporting its role in DNA repair and/or RNA transcription. It is likely that these large proteins will eventually be implicated in a variety of cellular processes, only some of which will be related to their role in the etiology of breast and ovarian cancer.
Mutations in BRCA1 and BRCA2
Over 600 mutations and sequence variations in BRCA1 have already been described. A large percentage of these lead to frameshifts resulting in missing or nonfunctional proteins, supporting the hypothesis that BRCA1 is a tumor suppressor gene. Like BRCA1, most of the BRCA2 mutations reported to date consist of frameshift deletions, insertions, or nonsense mutations leading to premature truncation of protein transcription.
The Role of BRCA1 and BRCA2 in Sporadic Cancer
Given that germline mutations in BRCA1 or BRCA2 lead to a very high probability of developing breast and/or ovarian cancer, it was a natural assumption that these genes would also be involved in the development of the more common nonhereditary forms of the disease. To date, only weak connections have been made between these genes and sporadic breast and ovarian cancer. Somatic mutations are absent in BRCA1, and a very low frequency of BRCA2 mutations exist in most breast cancer tumor tissue.
Molecular Correlations
Mutations in BRCA1 and BRCA2 confer a highly increased susceptibility to breast and ovarian cancer, but these mutations do not lead to cancer by themselves. The current consensus is that these are gatekeeper genes that, when removed, allow other genetic defects to accumulate. The nature of these other molecular events may define the pathway through which BRCA1 and BRCA2 function.
A number of studies have looked at steroid hormone receptor levels in tumors containing BRCA1 and BRCA2 mutations. In most of these studies, BRCA1 cancers were shown to be more often estrogen receptor-negative than nonhereditary cancers.
Background of genetic polymorphisms and Breast Cancer Risk
The search for genetic markers for breast cancer susceptibility has led to an
increasing number of epidemiologic studies of relatively common genetic
markers referred to as genetic polymorphisms. These polymorphically
expressed genes code for enzymes that may have a role in the metabolism
of estrogens or detoxification of drugs and environmental carcinogens.
Although the clinical significance and causality of associations with breast
cancer reported to date is unclear, genetic polymorphisms may account for
why some women are more sensitive than others to environmental
carcinogens such as replacement estrogens or cigarette smoke.
Implication of estrogen receptor in breast cancer
Estrogen receptor alpha (ER) plays a key role in the development and progression of
breast cancer as well as the treatment and outcome of breast cancer patients. Breast cancer can be classified in Estrogen receptor-positive breast cancer and estrogen
receptor-negative breast cancer.
The growth of estrogen receptor (ER)-positive breast
cancer cells is hormonally regulated, but the majority of breast cancers are ER negative
and unresponsive to hormonal therapy.
The estrogen receptor (ESR) is a ligand-activated transcription factor
composed of several domains important for hormone binding, DNA
binding, and activation of transcription.
Gene estructure
Human ESR gene is more than 140 kb long. It contains
8 exons, and the position of its introns has been highly conserved.
One mechanism suggested to play a role in the progression of human breast cancer from hormone
dependence to independence is the expression or altered expression of mutant and/or variant forms of
the estrogen receptor. Murphy et al. (1996) stated that 2 major types of variant ESR mRNA had been
reported in human breast biopsy samples so far: truncated transcripts and exon-deleted transcripts.
Murphy et al. (1996) provided data on a novel type of abnormal ESR mRNA. They found
larger-than-wildtype ESR mRNA RT-PCR products in 9.4% of 212 human breast tumors analyzed.
Cloning and sequencing of these larger RT-PCR products showed 3 different types: complete
duplication of exon 6 in 7.5%; complete duplication of exons 3 and 4 in 1 tumor; and a 69-bp insertion
between exons 5 and 6 in 3 tumors. While it is unknown if these novel ESR-like mRNAs are stably
translated in vivo, any resulting protein would be structurally altered, possibly resulting in altered
function.
Two isoforms of the human ESR, ESRA and ESR-beta (ESR2; 601663), occur, each with distinct
tissue and cell patterns of expression. Additional ESR isoforms, generated by alternative mRNA
splicing, have been defined in several tissues and are postulated to play a role in tumorigenesis or in
modulating the estrogen response
Using transient transfection assays, Fan et al. (1999) demonstrated that BRCA1 inhibits signaling by the
ligand-activated estrogen receptor ESR-alpha through the estrogen-responsive enhancer element and
blocks the C-terminal transcriptional activation function AF2 of ER-alpha. These results suggested that
wildtype BRCA1 protein may function, in part, to suppress estrogen-dependent mammary epithelial
proliferation by inhibiting ESR-alpha-mediated transcriptional pathways related to cell proliferation, and
that loss of this ability may contribute to tumorigenesis.
Walter et al. (1985) determined that the human ESR gene maps to chromosome 6. By in situ
hybridization, using a cDNA probe containing the coding sequence for the estrogen receptor, Gosden
et al. (1986) assigned the gene to 6q24-q27.
It is accepted that the presence of estrogen receptor identifies those breast cancer patients with a lower
risk of relapse and better overall survival (Clark and McGuire, 1988), and the measurement of ESR
has become a standard assay in the clinical management of breast cancer. Receptor status also
provides a guideline for those tumors that may be responsive to hormonal intervention. But only about
half of ESR-positive patients respond to various hormonal therapies and of those who do respond
initially, most will eventually develop hormonally unresponsive disease following a period of treatment
even though ESR is often still present. Sluyser and Mester (1985) hypothesized that the loss of
hormone dependence of certain breast tumors may be due to the presence of mutated or truncated
steroid receptors that activate transcription even in the absence of hormone. Fuqua et al. (1993)
reviewed ESR mutations that may be important in breast cancer progression. Scott et al. (1991), for
example, had found truncated forms of DNA-binding ESR in human breast cancer.
More information about BRCA1
More information about BRCA2
More information about RE
Microarray technique
One way to obtain useful information about a genome is to determine which genes are induced or repressed in response to a phase of the cell cycle, a developmental phase, or a response to the environemen, such as treatment with a hormone. Sets of genes whose expression rises and falls under the same condition are likeley to have a related function. In addition, a pattern of gene expression may also be an indicator of abnormal cellular regulation and is a useful tool in cancer diagnosis. Because genomes are so large,a new technology has been developed for studying the regulation of thousands of genes on a microscope slide.
Microarray analysis is a new technology in which we can study the expression of all or most of the genes of an organism. DNA microarrays are devices of small size (chip) that contain biological material (bio) and that are used for obtaining genetic information. The technique is based on the hybridation method of Southern but the unlabeled bimolecule is fixed to a solid support and is hybridized to the labeled samples. After hybridation and washes quantitative detecction of binded product has to be done.
This technique has lots of applications depending of which biomolecule is being used (cDNA, DNA or oligonucleotides). With microarrays can be studied and obtained information about many molecules at the same time.
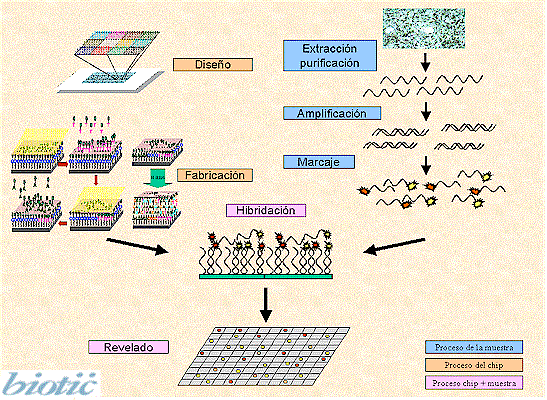

For more infromation you can consult Infobiochip
Summary of the article
Recently van't Veer et al ( Nature 415:530--536,2002) have published an article using DNA microarrays analysis in order to classify primary breast tumours and identify a gene expression signature strongly predictive of a short interval to distant metastases ('poor prognosis' signature) in patients without tumour cells in local lymph nodes at diagnosis (lymph node negative). The poor prognosis signature consists of genes regulating cell cycle, invasion, metastasis and angiogenesis. This gene expression profile will outperform all currently used clinical parameters in predicting disease outcome. The findings of the authors provide a strategy to select patients who would benefit from adjuvant therapy.
The article includes three different experiments:
(1) Method of unsupervised two-dimensional clustering to cluster and correlate 98 tumours and 25000 genes.
(2) Method of supervised classification of breast cancers into prognosis or diagnostic categories based on gene expression profiles.
(3) Supervised two-layer classification.
(1) They have studied the differential expression of 25000 genes in 98 primary breast cancers selected: 34 from patients who developed distant metastases within 5 years (LT5Y), 44 from patients who continued to be disease-free after a period of at least 5 years (GT5Y), 18 from patients with BRCA1 germline mutations, and 2 from BRCA2 carriers. All 'sporadic' patients were lymph node negative, and under 55 years of age at diagnosis.
To apply the microarray technique, 5 microg total RNA was isolated from snap-frozen tumour material from each patient and used to derive complementary RNA (cRNA). A reference cRNA pool was made by pooling equal amounts of cRNA from each of the sporadic carcinomas. Two hybridizations were carried out for each tumour using a fluorescent dye reversal technique on microarrays containing approximately 25000 human genes synthesized by inkjet technology13. Fluorescence intensities of scanned images were quantified, normalized and corrected to yield the transcript abundance of a gene as an intensity ratio with respect to that of the signal of the reference pool14.
Some 5000 genes were significantly regulated across the group of samples (that is, at least a twofold difference and a P-value of less than 0.01 in more than five tumours). It is important to specify that when the authors consider 'less than' they mean 'less or equal than'.
In the two-dimensional cluster analysis (method of unsupervised two-dimensional clustering), gene clustering and tumour clustering were performed independently using an aglomerative hierarchical clustering algorithm. For gene clustering, pairwise similarity metrics among genes were calculated on the basis of expression ratio measurements across all tumours. Similarly, for tumour clustering, pairwise similarity measures among tumours were calculated based on expression ratio measurements across all significant genes (for details, the linkof the authors has been attached, Suplementari Information).
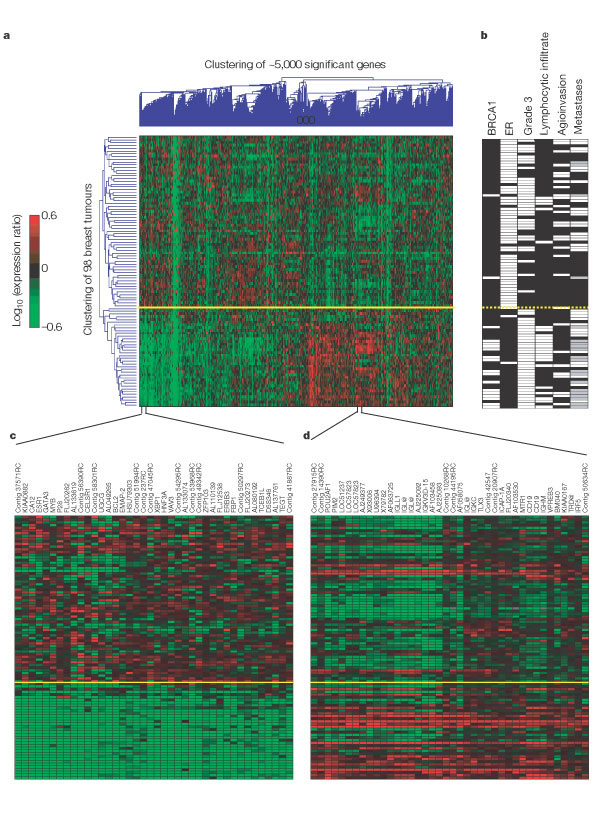
This method of clustering allowed the authors to cluster the 98 tumours on the basis of their similarities measured over these approximately 5000 significant genes, and similarly, to cluster the 5000 genes on the basis of their similarities measured over the group of 98 tumors (Fig.1a).
The lenght and the subdivision of the branches of the dendogram resulting from the cluster analysis, represents the proximity and relatedness of the tumours (left side of the dendogram or y-axes), and the relatedness of the expression of the genes (top of the dendogram or x-axes). Each row (y-axes) represents a tumour and each column single gene (x-axes).
The colors stand for: red indicates upregulation, green downregulation, black no change and grey no data available. The yelow line marks the subdivision into two dominant tumour clusters.
Two distinct groups of tumours (y-axes) are the dominant feature in this two-dimensional display (top of plot representing 62 tumours and bottom of plot representing 36 tumours), suggesting that the tumours can be divided into two types on the basis of the expression of this set of 5000 significant genes. In the upper group of 62 tumours,they found that 34% of the sporadic patients were from the group who developed distant metastases within 5 years, whereas in the lower group of 36 tumours, 70% of the sporadic patients had progressive disease, this is they developed metastases in less than 5 years (Fig. 1a).
From these percentages we infer that: the upper group of tumours could be associated with a 'good prognosis', and although, the genes that correlate with them could be predictors of their prognosis; the other way around, the lower group of tumours could be associated with a 'poor prognosis' and the genes that correlate could be used as predictors of 'poor prognosis'. Thus, using unsupervised clustering they can already, to some extent, distinguish between 'good prognosis' and 'poor prognosis' tumours.
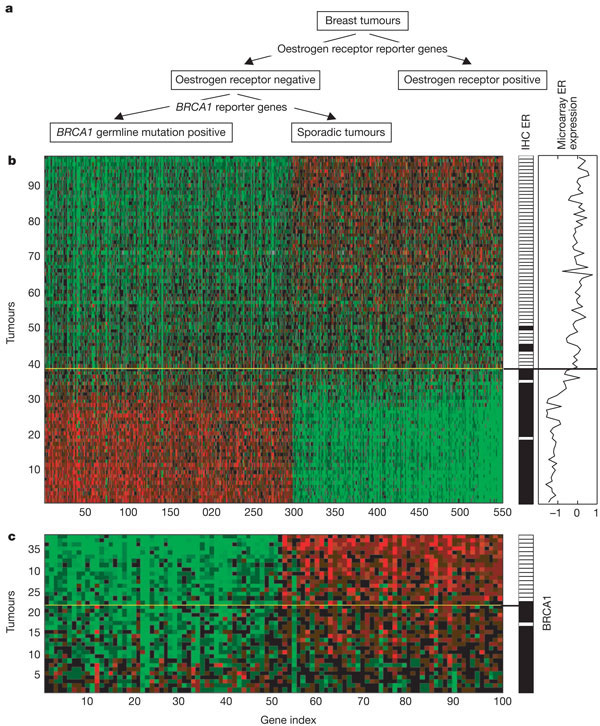
To gain extra information of the genes of the dominant expression signatures, the authors associated them with histopathological data. The figure 1b contains the results: white indicates positive, black negative and grey denotes tumors derived from BRCA1 germlines carriers who were excluded from the metastasis evaluation.
- The first column represents the BRCA1 germline mutation carriers versus sporadic patients. They observed that out of 18 patients with BRCA1 mutations, 16 of them were clustered together in the bottom branch of the tumor dendogram (containing 36 tumors).
- They studied the expression of estrogen receptor (ER) alfa, which had been determined by immunohistochemical (IHC) staining.
They obtained 39 tumours with negative expression for ER-alfa, (they had a mutated and non functional receptor for estrogens that would not respond to hormonal therapy). They observed that 34 of them clustered together in the bottom branch of the tumour dendrogram (containing 36 tumors), which , in our opinion, could be associated with 'poor prognosis' tumours.
In the figure 1c, the y-axes represents the 98 tumors studied and the x-axes a set of genes containing both the ER gene (ESR1) and genes that are apparently co-regulated with ER (some of which are known ER target genes). We observe that the 36 tumours with 'poor prognosis' (bottom of the dendogram) coincide with a group of downregulated genes (green).
- They classified the tumours in three different grades (1,2,3). In the third column of figure 1b they represent the grade 3 tumours in white (positive) and the oder grades in black (negative). We can observe that 35 out of the 36 tumours of the bottom of the dendogram, which are associated with a 'poor prognosis', are grade 3 tumours. Although, there are many grade 3 tumours in the upper cluster too, they concluded that the grade tumour is not a specific feature of either of the clusters.
- The fourth column represents the lymphocytic infiltration, and it seems that the 36 tumours with 'poor prognosis' show a positive lymphocytic infiltration.
The figure 1d represents an enlarged portion of the dendogram in figure 1a, were they represent the tumours and their correlation with a set of genes expressed primarily by B and T cells. From the figure 1d we infer that the second dominant gene cluster (bottom of the dendogram) is associated with lymphocytic infiltration because those tumours upregulate (red) with the chosen genes.
- Clinical data about the angioinvasion of the 98 tumours is represented in the fifth column. The information that we can extract is not as significant as the information from the previous columns because the positives and the negatives are not so correlated with the two subdivisions of the tumours. They conclude that the angioinvasion is not a specific feature of either of the clusters.
- The last column contains clinical data about the metastases of the tumours, and as expected, 30 out of 36 tumors with 'poor prognosis' appear with metastases.
First of all, they concluded that most BRCA1 mutant tumours are ER negative and manifest a higher amount of lymphocytic infiltration. They also confirm the fact that the gen BRCA2 is not implicated in inherited breast cancer, as the two tumours of BRCA2 carriers are part of the upper cluster of tumours and do not show similarity with BRCA1 tumours. The final conclusion is that the unsupervised clustering detects two subgroups of breast cancers, which differ in ER status and lymphocytic infiltration.
(2) They were not convinced enough of the results obtained with the unsupervised two-dimensional clustering, so in order to identify reliable 'good and poor prognosis' tumours they decided to use a powerful three-step supervised classification method. This method classifies breast tumours into prognosis and diagnostic categories based on gene expression profiles, and it includes the following three steps:
- Selection of discriminating candidate genes by their correlation with the category (LT5Y and GT5Y);
- Determination of the optimal set of reporter genes using a leave-one-out cross validation procedure;
- Prognostic (diagnostic prediction) based on the gene expression of the optimal set of reporter genes.
(for details see Suplementari Information)
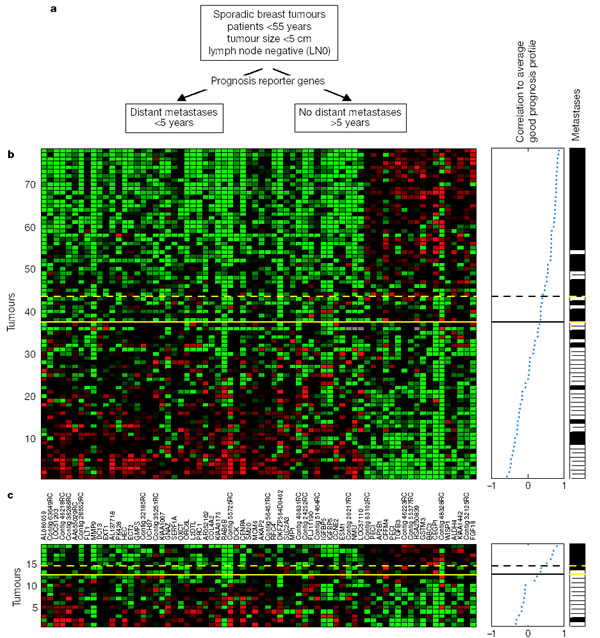
They selected 78 sporadic lymph-node-negative patients (these number comes from the original 98 patients without the 20 BRCA carriers) to search specifically for a prognosis signature in their gene expression profiles: 44 patients remained free of disease after their initial diagnosis for an interval of at least 5 years (good prognosis group or GT5Y) and did not develop metastases until an average of 8.7 years, and 34 patients had developed distant metastases within 5 years (poor prognosis group or LT5Y), the mean time for metastases in this group is 2.5 years (Fig. 2a).
Again, approximately 5000 genes significantly regulated were selected from the 25000 genes on the microarray, those 5000 are not necessairly the same 5000 genes used in the previous experiment.
The paramaters for these selection are at least a twofold difference (the logarithm basis 10 of the ratio of expression is equal to a correlation coefficient <-0.3 or >0.3) and a P-value of less than 0.01 in more than 3 tumours out of 78.
There were found 231 genes to be significantly associated with disease outcome. We can find those 231 genes in the table S2 from Suplementari Information or in the file prognosisrepgenes.txt
The next step is to rank-order these 231 genes on the basis of the magnitude of the correlation coefficient.
Finally, the number of genes 'prognosis' selected and classied was optimized by sequentially adding subsets of 5 genes from the top of this rank-ordered list and evaluating its power for correct classification using the 'leave-one-out' method for cross-validation (see Suplementari Information).
Classification was made on the basis of the correlations of the expression profile of the 'leave-one-out' sample with the mean expression levels of the remaining samples from the good and the poor prognosis patients, respectively.
The main point of this supervised method is that the clustering is done taking into account the previous information known about the tumours, which allowed the investigators to select the information a priori before the cluster is done. Thus, the results of the clustering are more significant and are a better approach to reality. They continued with this method improving the accuracy until the optimal number of marker genes was reached. They obtained a final group of 70 genes which were strong predictors of the disease outcome.
The expression pattern of the 70 genes is shown in the colour plot of Fig.2b. They represent the expression data matrix of those 70 prognosis marker genes from tumors of 78 patients, each row represents a tumour and each column a gene.
As said before, gens are ordered according to their correlation coefficient with the two prognosis groups, and tumours are ordered by the correlation to the average profile of expression of the good prognosis group.
After the evaluation of the results, we conclude that the tumours above the yelow line are 'good prognosis'and the ones below are 'poor prognosis' tumours. The 'poor' prognosis reporter genes in the left side of the matrix, downregulate (green) with the 'good prognosis' tumours and upregulate (red) with the 'poor prognosis' tumours.
On the other hand, the 'good prognosis' reporter genes, upregulate (red) with the 'good prognosis' tumours and downregulate (green) with the 'poor prognosis' tumours.
The middle panel of the figure represents the average profile of expression of the good prognosis group (represented between -1 and 1) and the correlation between each tumour and these average. The solid line in this panel represents the prognostic classifier with optimal accuracy, and the dashed line, the prognosis classifier with optimized sensitivity.
Above the dashed line, patients have a good prognosis signature, and below, a poor signature, though, the tumours are subdivided by the yelow line in good and poor prognosis signature.
The column in the right side of the figure represents the metastases status for each patient: white indicates patients who developed distant metastases within 5 years after the primary diagnosis, black indicates patients who continued to be disease-free for at least 5 years.
This classification coincides with the results of the middle pannel and with the regulation of the different reporter gens within the 98 tumours represented in the left panel (because we find more metastases (white) in the bottom of the column), which coincides with the 'poor prognosis' tumours.
After evaluating the results ofthe three panels, the authors concluded that the classifier predicted correctly the actual outcome of disease for 65 out of the 78 patients, which is an 83%.
We can see the 17% left, thus the prognosis patients assigned to the opposite category, in the right panel of Fig.2b were there are 5 positive (white) tumours for metastases within the predicted 'good prognosis' tumours, and 8 negative (black )tumours for metastases within the predicted 'poor pognosis' tumours.
However, the authors concluded that to select of the appropriate patients for adjuvant systemic therapy, a lower number of poor prognosis patients assigned to the 'good prognosis' category should be attained.
To that purpose, they set new parameters of selection, establishing a threshold that resulted in another expression pattern colour plot with a wrong classification of no more than 10% of the poor prognosis patients (3 patients out of 34 of the poor prognosis group were not correctly classified).
This optimized sensitivity threshold resulted in a total of 15 misclassifications: 3 poor prognosis tumours were classified as good prognosis, and 12 good prognosis tumours were classified as poor prognosis. They even found small primary tumours without lymph node metastases classified by this method as ' poor prognosis' signature, indicating that those genes are already programmed for this metastatic phenotype.
The functional annotation for the selected genes provided more information into the underlying biological mechanism leading to rappid metastases.
The gene names in Fig.2b demonstrate that genes involved in cell cycle, invasion and metastasis, angiogenesis, and signal transduction are significantly upregulated (red) in the poor prognosis signature. When they evaluated all 231 prognostic reporter genes, more genes belonging to these functional categories became apparent (Table S2 of Suplementari Information ).
They noticed that many clinical studies had correlated alterations in expression of individual genes with breast cancer disease outcome, often with contradictory results. Some examples of the genes correlated with the outcome of the pathology in those clinical trials are cyclin D1, ER alfa,UPE, PAI-1, Her2/neu (cerbb2) and c-myc.The surprising fact is that none of these genes were present in their set of 70 marker genes selected by the supervised method, while they expected that their 70 reporter genes were strong predictors of the prognosis of breast cancer. They attributed these contradiction to the fact that they were determining gene expression at the level of transcription, whereas most previous studies measured protein levels. However, they concluded that the isolated genes from the clinical trials had only limited predictive power, which highlighted the meaning of their choice: to carry out an approach to the genes predictors based on the analysis of many genes.
To validate the prognosis classifier (set of 70 genes selected by the supervised method), an additional independent set of primary tumours from 19 young, lymph-nodenegative breast cancer patients was selected. This group consisted of 7 patients who remained metastasis free for at least five years (GT5Y), and 12 patients who developed distant metastases within five years (LT5Y).
The disease outcome was predicted by the 70-gene classifier and the results showed in Fig.2c give 2 out of 19 incorrect classifications using both the optimal accuracy threshold (solid line) and the optimized sensitivity threshold (dashed line). Thus, the classifier showed a comparable performance on the validation set of 19 independent sporadic tumours and confirmed the predictive power and robustness of prognosis classification using the 70 optimal marker genes.
They developed many statistical test, and finally they conclude that in order to provide more accurate estimation of the metastatic risk associated with the prognosis signature, it is necessary to study a large and unselected group of breast cancer patients.
(3) To investigate the expression patterns associated with the immunohistochemical staining of ER (positive or negative) and to explore the differences between the sporadic tumours and the BRCA1 tumours that cluster within the ER negative tumours (as they showed in Fig.1), they performed a supervised two-layer classification which is shown in Fig.3a.
They classified 98 breast tumours into an ER positive group and an ER negative group, which was further divided into BRCA1 mutation or sporadic tumours.
Figure 3b shows the expression data matrix of the 98 tumours across 550 optimal ER reporter genes (see Suplementari Information Table S3) classified on the basis of their level of contribution to the classifiers. The colour patterns discriminate between tumours with an ER negative signature (below the yelow line) and ER positive tumours (above the yelow line).
The right panel of ER status determined by immunohistochemical data, indicates that only 2 tumours are classified with the microarray data as ER negative tumours while there are positive, and that three negatives ones are placed in the positive side. They attempted 95% of correct classification. Thus, they concluded that the observation in the unsupervised analysis (Fig.1) that ER clustering had predictive power for prognosis is also valid for the ER supervised classification (Fig.3), although the ER status does not reach the level of significance of the prognosis classifier (70 reporter genes).
In the figure 3c we observe the expression data matrix of 38 ER negative tumours (y-axes) and 100 optimal BRCA1 reporter genes (x-axes). (The data is available for downloading in the Suplementari Information Table S4). Patients above the yelow line are characterized by a BRCA1 signature. The classification into sporadic and BRCA1 tumours is mainly due to the differences in levels of gene expression (amplitude). We can easily see the tumours divided into two subgroups: BRCA1-like tumours and sporadic tumours.
The ER negative and BRCA1 positive tumours appear in the top-right side of the plot and upregulate (red) with the BRCA1 reporter genes. The ER negative and sporadic tumours appear in the top-left side of the plot and downregulate (green).
This results coincide with the panel that indicates the satus of the BRCA1 germline mutation (white indicates positive and black indicates negative or sporadic tumour). The accurancy of the analysis was of 95%, with only one positive BRCA1 tumour classified in the bottom of the column as negative. The one sporadic tumour that was classified as a BRCA1 tumour was shown to contain methylation of the BRCA1 promoter, indicating an epigenetic modification of BRCA124 (data not shown). Thus the discordant BRCA1 tumour is from a patient where the germline mutation had only altered the last 29 amino acids of the BRCA1 protein (BRCA1 mutation 5,622del62) ( which abolishes transcriptional activation by BRCA125). This argument explains that although the cancer was BRCA1 positive, it was classified as negative because the methylation abolished the trasncriptional activation.
From the results obtained, the authors conclude that breast cancer prognosis can already be derived from the gene expression profile of the primary tumour. The purpose of their article was to find strong predictors of the prognosis of the disease in order to select which patients would benefit from the adjuvant therapy, and to avoid these therapy to those patients that may not benefit from it. The problem could be that the predictors selected would not be restrictive enough and give false negative (determine that a patient would not benefit from the treatment when he really would need the adjuvant therapy). Table 1 in the article shows that the prognosis classifier selects just as effectively those high-risk patients that would benefit from adjuvant therapy, but significantly reduces the number of patients that receive unnecessary treatment. Thus, the prognostic profile potentially provides a powerful tool to tailor adjuvant systemic treatment that could greatly reduce the cost of breast cancer treatment, both in terms of adverse side effects and health care expenditure. Furthermore, the signature that defines ER
status can be used to decide on adjuvant hormonal therapy, and the signature that reveals BRCA1 status may further improve the diagnosis of hereditary breast cancer .
Finally, genes that are overexpressed (red) in tumours with a 'poor prognosis' profile are potential targets for the rational
development of new cancer drugs. Identification of such targets may improve the efficiency of developing therapeutics for many tumour types.
Our project
INTRODUCTION
It is known that breast cancer patients with the same stage of disease can have markedly different treatment responses and overall outcome.
It is hard to classify accurately breast tumours according to their clinical behaviour, even with the strongest predictors for metastases, for example, lymph node status, histological grade and immunohistochemical techniques. The clinical experience demonstrates that chemotherapy or hormonal therapy reduces the risk of distant metastases by approximately one-third; however, 70-80% of patients receiving this treatment did not respond to the therapy. Those patients could though avoid the pain, the adverse side effects and the incremented risk of metastases of chemotherapy or hormonal therapy, and the cost of breast cancer treatment for the Health care. None of the signatures of breast cancer gene expression reported to date, allow for a priori prediction of which patients need those kind of treatments.
OBJECTIVES
From the first figure in the article, the authors deduce that there are a group of genes that correlate with the prognosis of the pathology. A specific genic expression pattern could avoid false positive diagnostic in the need of chemiotherapy.
They distinguish a group of genes that are correlated with a good prognosis of cancer, this is that metastases do not appear during the first five years (GT5Y); and a group of genes that are correlated with a bad prognosis (LT5Y), this is the developement of metastases within the first five years. We thought that the criteria used by the authors deciding which gene is significantly regulated, this is at least a twofold difference in the ratio of expression and a Pvalue of less than 0.01 in more than five tumors, was not restrictive enough, and though, the correlation found could not be enough significant. We could not conclude that those genes were really strong predictors of the prognosis of the cancer.
The main point of our project is to establish more restrictive parameters to select among the 25000 genes, those that are best correlated with the prognosis of the 98 tumors studied. The result that we expect is to clearly find two separate groups of genes that strongly correlate with disease outcome and that are better predictors than the ones defined by the authors.
MATERIALS and METHODS
We began with a file that contained the group of 4968 genes that the authors concluded that correlated with the prognosis of the pathology (Fig.1). Initial file
It would have been better to start up from the original 25000 genes that the authors selected for their experiments, but unfortunately we had problems with the memory of our computers and we could not deal with such a big file. As we establish in the objectives, we needed to define more restrictive parameters to select among the 5000 genes the best predictors. We developed a program with LINUX system (using Perl lenguage) that allows the user to select from the inital file only those genes that fulfill the parameters chosen by the user. We did the program in an interactive way, in order to make it easy to select the parameters and avoid the program to abbort due to compilation errors. The program can be found in gens.pl .
The program gens.pl allows the user to select the following parameters:
- Logaritm (base 10) of the Ratio
- Pvalue
- Minimum number of tumours
- File for working
- File for saving results
The logaritm of the Ratio is the mean ratio of the red and green channels for a given probe on the chip. High quality data are derived from the genes associated with the greatest signal intensity. This reflects the extend of induction or repression of a given gene. If the user works with the log10, a mean ratio of 100 means that the gene was induced 100 fold by the perturbation. A mean ratio of 0.01 means that the genes repressed 100 fold.
The P-value is the confidence level that a mean ratio of a gene is significantly different from 1, or no change. A P-value of 3.2E-03, for example, exceeds the 99% confidence (P-value = 0.01).
The minimum number of tumours means the minimum number of tumours in which the gene must have the established parameters (P-value and Log10 of the Ratio) to be selected.
The program asks the user on which file containing the data, the restrictive parameters have to be applied.
Finally the user can choose in which file he wants to save the outfile containing only the selected data.
Our group ran this program to select from the file that contained the 4968 genes of the authors different files using different parameters of restriction. All the files that we created from the original one are detailed on the Table 1 below. To import data to this program, it must have a particular format, described below.