Un ejemplo de este tipo de estudios lo encontramos en el articulo Gene expression profiling predicts clinical outcome of breastcancer publicado en Nature (Nature, vol.415, 530-535) en Enero del 2002. En éste, se intentó encontrar mediante la técnica de microarray de DNA un set de genes que permitiera predecir el desarrollo de cáncer de mama (uno de los que tiene mayor incidencia mundial) , causando cerca de trescientasmil muertes cada año.
Basándose en este
artículo se ha intentado comprobar si un nuevo set de genes con
distintas condiciones de expresión sería igualmente predictivo.
Para esto se creó un programa en lenguaje Perl (geneselection.pl)
que permitía seleccionar de un grupo de genes sólo aquellos
que cumplían unas determinadas condiciones de expresión.
Los resultados fueron procesados utilizando dos programas, Cluster(para
el cálculo de distancias de expresión) y TreeView(para
su visualización).
Se ha visto
que sólo entre un 20% y un 30 % de los tratamientos con quimioterapia
en mujeres con cáncer primario de mama eran realmente necesarios,
ya que el resto de casos hubieran podido sobrevivir sin él. He aquí
la necesidad de tener un set de buenos predictores genéticos .
Para encontrar el set de genes se analizó el DNA de 117 mujeres con cáncer primario de mama, de las cuales se seleccionaron 98: 78 esporádicos ( 34 desarrollaron metástasis en menos de 5 años y 44 tardaron más), y el resto no esporádico o hereditario ( mutación en el gen de BRCA1 o BRCA2). De cada tumor se analizaron por DNA microarray 25,000 genes obtenidos de dos base de datos:( Ref Seq y ESTs) También se incluyeron unos 1,200 genes que no tenían relación con el cáncer para utilizarlos como control. De estos genes se obtuvieron los oligos que se utilizarían para hacer dos hibridaciones con las muestras problema.
Se extrajo 5 microgramos de RNA de cada uno de los tumores obteniéndose su cRNA mediante transcripción in vitro y se marcó con fluorescencia (CyDye). A partir de estas muestras se creó un pool que contenía cantidades iguales de los tumores esporádicos para obtener un valor de expresión de referencia.
Tras la hibridación se quantificó la fluorescencia por microscopia láser confocal obteniéndose un valor de expresión que se utilizó para calcular el ratio de expresión (expresión gen/expresión referencia).
Del conjunto total, 5,000 genes cumplían las siguientes condiciones de expresión:
· Pvalue menor o igual al 0.01.
· Una expresión dos veces
mayor o dos veces menor que el valor de referencia (ratio de expresión)
· Que las dos condiciones anteriores
se cumplan como mínimo en 5 tumores.
Para ver la relación
de expresión entre estos 5,000 genes se realizó un algoritmo
de agrupamiento (o clustering) jerárquico no supervisado, que también
se aplicó para encontrar similitudes entre los 98 tumores. A partir
de este clustering se delimitaron dos grupos diferenciados (imagen
1a).
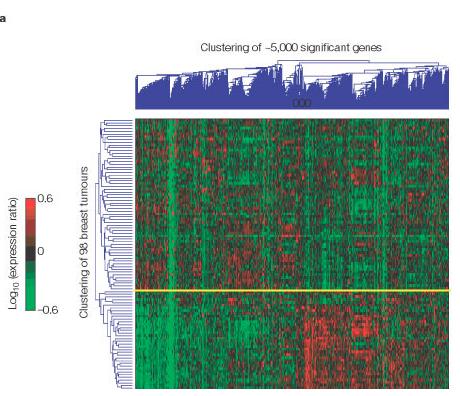
(*)Imagen
1a: el grupo superior (prognosis favorable) corresponde
a 62 tumores, la mayoría de los cuales (66%) no desarrollaron metástasis
en menos de 5 años; mientras que el grupo inferior (prognosis
desfavorable) corresponde a 36 tumores, la mayoría de los cuales
(70%) desarrollaron metástasis.
Además se buscaron parámetros clínicos asociados al cáncer (expresión receptor de estógenos ER, mutaciones en la línea germinal de BRCA1, infiltración linfocítica, angioinvasión, y grado del tumor) (imagen 1b).

(*)Imagen
1b: se
observó que en el grupo de prognosis favorable había
menor infiltración linfocítica, mayor expresión del
gen de ER-alfa y no presentaba mutación en el gen BRCA1, mientras
que en el grupo de prognosis desfavorable el patrón es opuesto
(el negro significa expresión negativa y el blanco expresión
positiva)
En los resultados de este
primer análisis se observa un cierto error en el pronóstico
de las pacientes. Con tal de mejorarlo, se seleccionan otros 5,000
genes de entre los 25,000 iniciales, siguiendo un método parecido
al anterior, pero esta vez el gen únicamente debía expresarse
en tres o más tumores esporádicos. Seguidamente se calcularon
los coeficientes de correlación de cada uno de estos genes, destacando
231 por tener una correlación altamente significativa con el desarrollo
de la enfermedad (más de 0.3 o menos de -0.3). Se ordenaron en virtud
a una mayor o menor correlación y se fueron añadiendo en
grupos de 5 genes, esta vez utilizando un método de clustering supervisado
(leave-one-out), al set de 5000 genes predictivos iniciales, con
tal de optimizarlo (imagen 2b).
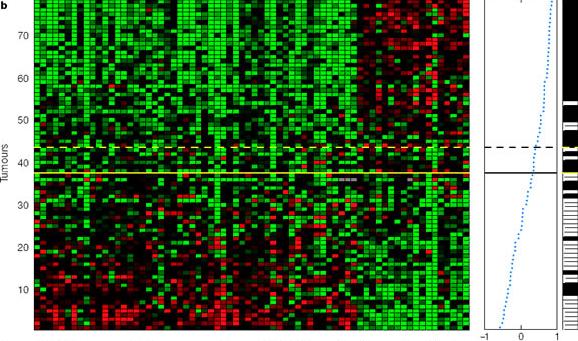
(*)
Imagen
2b: en el panel izquierdo se observan los 78 tumores divididos según
su prognosis. En el panel de la derecha se muestra la tendencia de correlación
(línea de puntos azules), así como la aparición de
metástasis (barras blancas).
Así se obtuvo un set definitivo de 70 genes que establecía un umbral de exactitud (línea contínua en el panel izquierdo de la imagen 2b) que permitía diferenciar entre el grupo de prognosis favorable (44 tumores) y desfavorable (34 tumores). A pesar de la eficacia del método de clasificación 13 de los 78 tumores se clasificaron de forma incorrecta (sensibilidad del 83%), por lo que se definió un umbral de sensibilidad (línea discontínua del panel izquierdo de la imagen 2b) que permitía un error menor al 10%.
Esta selección de 70 genes fue contrastada con resultados obtenidos por otros grupos, viéndose que ninguno de nuestros genes se incluía en dichos resultados. La explicación la podríamos encontrar en el hecho de que los estudios anteriores se basaban en el nivel proteíco mientras que en éste se analizó la expresión del gen.
Este programa nos
selecciona , dado un fichero de genes que se expresan en tumores, aquellos
que cumplan una serie de condiciones : nivel de expresión (calculado
como log10
del ratio), p-value y número mínimo
de tumores en los que se expresan. De esta manera podemos obtener ficheros
con subconjuntos de genes que suponemos más o menos relacionados
con el desarrollo de la enfermedad.
2- Genes que se expresan como mínimo en 30 tumores con un ratio
de expresión mayor o igual a 2.
* genes predictivos BRCA1: cwbrca30tr2.txt
* genes predictivos ER: cwer30tr2.txt
* genes predictivos prognosis: cwpr30tr2.txt
Para hacer las selecciones
variamos dos condiciones de expresión: el ratio (2 y 3) y el número
de tumores (5, 15 y 30). Comprobamos que una variación del ratio
tiene mayor repercusión en el número de genes que una
variación en el número de tumores expresados (tabla
2).
Al restringir las condiciones
de expresión, el número de genes del fichero inicial
disminuye,así como el número de genes
BRCA1, ER y prognosis. Si el método de selección fuera bueno
el número de genes de BRCA1, ER y prognosis se mantendría
mientras que el resto disminuiría, lo que significa que su
porcentaje debería aumentar en lugar de conservarse (tabla
3). Esto nos permite concluir que unas
condiciones de expresión muy estrictas originan una pérdida
importante de información, para obtener un diagnóstico correcto
de la evolución de la enfermedad se necesita analizar un cantidad
mayor de genes.
Primeramente, analizamos los genes BRCA1,ER
y prognosis
coincidentes con el fichero inicial. En los tres árboles
observamos dos ramas principales que corresponden a dos grupos diferenciados
de tumores según su patrón de expresión génica
(en rojo, los genes sobreregulados y en verde los infraregulados). Si los
genes tienen poder predictivo, las dos ramas separaran el grupo de prognosis
favorable ("greater than 5 years") del de prognosis desfavorable (BRCA1
y "less than 5 years".




En segundo lugar,
compararemos los microarrays de un mismo grupo de genes en diferentes condiciones
(en este caso, intentaremos ver si variando el número mínimo
de tumores, las diferencias son mayores).
Basándonos
en las tablas de porcentaje y en el análisis de los microarrays
concluimos:
A) Que los tumores se agrupan en un grupo relacionado con una prognosis favorable y otro con una prognosis desfavorable.
B) Que las condiciones astringentes reducen en un porcentaje importante el número de genes de nuestros sets predictivos, por lo que su valor en el diagnóstico del cáncer es bastante limitado.
C) Que se aprecian diferencias en la construcción
de los árboles en medida del set predictivo utilizado, siendo el
grupo prognosis el menos sensible a los cambios
de condiciones.

Página
creada por:
![]() ÒNIA
ÒNIA ![]() AGRISTÀ
AGRISTÀ
![]() AQUEL
AQUEL ![]() ODRIGUEZ
ODRIGUEZ
![]() SABEL
SABEL ![]() ORENZO
ORENZO
![]() ONTSE
ONTSE ![]() UEVAS
UEVAS