La familia de proteínas Bcl2 se encuentra conservada
en todos los animales y está implicada en la alteración de la permeabilidad de
la membrana mitocondrial que resulta con la pérdida del citocromo C y la
activación de la apoptosis Según los datos experimentales, Ced9 de C.elegans (Cizeau
et al.),
el cluster homólogo a Bcl2 de poríferas (Wiens
M. et al.)
y Drosophila (Zhang
H. et al.)
son versiones pro-apoptóticas. Según Aravind
L. et al. la diferenciación entre las subfamilias pro y
anti-apoptóticas debe haber surgido con la aparición de los celomados ya que
uno de sus ancestros ya contenía una proteína con función anti-apoptótica. Ya en vertebrados podemos observar una gran
diversificación de las dos subfamilias, sobretodo en la pro-apoptótica. Esta
tendencia parece más importante en la subfamilia BH3 en la que se han
identificado muchas con baja similitud entre ellas sobretodo en mamíferos. Por
esto no queda muy claro si el dominio BH3 es funcionalmente relevante y si
tienen un único ancestro común. Ha sido ésta la razón por la que hemos
descartado utilizar esta subfamilia para nuestro análisis filogenético.
En vertebrados se observa un crecimiento en
la complejidad de les proteínas y los dominios asociados a la apoptosis. En el
caso de la familia de Bcl2 se debe principalmente a la duplicación de genes y a
la emergencia de nuevos dominios mediante la modificación de los preexistentes
o la reorganización de los mismos. Según Aravind
L. et al las innovaciones
en vertebrados en la señalización apoptótica y citoquinas relacionadas está
unida a la evolución del sistema inmunitario y más en concreto a la aparición
de nuevas líneas celulares que necesitan de vías reguladoras más
especializadas.
El misterio más grande en la evolución de la
apoptosis es la presencia de homólogos de la maquinaria reguladora en bacterias
(Frade
JM. et al.).
El hecho que actinomicetos y cianobacterias codifiquen para la mayoría de
dominios asociados a la apoptosis de la misma manera que lo hacen los
eucariotas contrasta con su ausencia en otros tipos de bacterias como las
arqueas. Esto sugiere la adquisición de estos genes por transferencia
horizontal, más probable de bacterias a eucariotas que no al revés.
En conclusión podemos
decir que la evolución del sistema apoptótico tiende al aumento de la
complejidad en vertebrados en comparación con nemátodos e insectos y se
manifiesta en el aumento de proteínas relacionadas con este proceso. Para entender el origen y la
evolución de la apoptosis se habrían de tener más genomas seqüenciados de las
diferentes ramas de la vida: cianobacterias, actinomicetos, eucariotas y
cordados primitivos.
Proteína
|
BH1 |
BH2 |
BH3 |
BH4 |
TM
|
||
|
Bak 2 Humana |
117-136 |
169-184 |
74- 88 |
- |
188-205 (potencial) |
||
|
Bak |
Humana |
117-136 |
169-184 |
74-88 |
- |
188-205 (potencial) |
|
|
Ratón |
114-133 |
166-181 |
71-85 |
- |
185-202 (potencial) |
||
|
BHRF1 Epstein-barr
virus |
89-109 |
142-157 |
- |
- |
166-186 (potencial) |
||
|
Bax-a
|
Bovino |
98-118 |
150-165 |
59-73 |
- |
172-192 (potencial) |
|
|
Humano |
98-118 |
150-165 |
59-73 |
- |
172-192 (potencial) |
||
|
Rata |
98-118 |
150-165 |
59-73 |
- |
172-192 (potencial) |
||
|
Ratón |
98-118 |
150-165 |
59-73 |
- |
172-192 (potencial) |
||
Bax-b
Humano
|
98-118 |
150-165 |
59-73 |
- |
- |
||
|
Bax-d Humano |
49-69 |
101-116 |
- |
- |
- |
||
|
Bcl2 |
Bovino |
126-145 |
177-192 |
83-97 |
10-30 |
202-223 (potencial) |
|
|
Hámster |
133-152 |
184-199 |
90-104 |
10-30 |
209-230 (potencial) |
||
|
a Humano |
136-155 |
187-202 |
93-107 |
10-30 |
212-233 (potencial) |
||
|
b humano |
136-155 |
187-202 |
93-107 |
10-30 |
196-239 (potencial) |
||
Pollo
|
130-149 |
181-196 |
87-101 |
10-30 |
208-228 (potencial) |
||
|
Rata |
133-152 |
184-199 |
90-104 |
10-30 |
209-230 (potencial) |
||
|
Ratón |
133-154 |
184-199 |
90-104 |
10-30 |
209-230 (potencial) |
||
|
Bcl-w |
Humano
|
85-104 |
136-151 |
- |
9-29 |
|
|
|
Ratón |
85-104 |
136-151 |
- |
9-29 |
- |
||
|
Bcl-x |
Humano |
129-148 |
180-195 |
86-100 |
4-24 |
210-226 (potencial) |
|
|
Pollo |
125-144 |
176-191 |
82-96 |
4-24 |
206-223 (potencial) |
||
|
Cerdo |
129-148 |
180-195 |
86-100 |
4-24 |
210-226 (potencial) |
||
|
Rata |
129-148 |
180-195 |
86-100 |
4-24 |
210-226 (potencial) |
||
|
Ratón |
129-148 |
180-195 |
86-100 |
4-24 |
210-226 (potencial) |
||
|
Bfl-1 (A1) |
Humano
|
77-97 |
132-147 |
- |
- |
- |
|
|
Ratón |
77-97 |
132-147 |
- |
- |
- |
||
|
Mcl-1 Humano |
252-272 |
304-319 |
209-223 |
- |
330-349 (potencial) |
||
|
CED9 Caenorhabditis briggsae |
159-179 |
213-228 |
- |
80-99 |
- |
||
CED9 Caenorhabditis elegans
|
160-179 |
213-229 |
- |
80-99 |
- |
||
|
Drob1 Drosophila melanogaster |
- |
- |
presente |
- |
- |
||
|
Lmh-5w African
swine fever virus |
76-95 |
126-141 |
- |
- |
1-18 (potencial) |
||
BHP1 Geodia cydonium |
presente |
presente |
- |
- |
- |
||
|
BHP1 Suberites
domuncula |
presente |
presente |
- |
- |
- |
||
Para realizar el estudio
filogenético hemos escogido los homólogos que hemos considerado como más
representativos. Hemos utilizado Bcl2 y Bclw tanto de humano como de ratón para
saber si se han generado antes o después de la radiación de los vertebrados y
Bfl1 como miembro lejano de la familia para saber si la familia entera ya
existía antes o después del mismo período.
Como homólogos, se han utilizado: Drob1
procedente de Drosophila melanogaster (Zhang
H. et al.), Ced9 de Caenorhabditis
elegans (Cizeau
J. et al.), Bhp1 de Geodia cydonium
(esponja) (Wiens
M. et al.) y Lmh-5w del virus africano de la fiebre porcina (Neilan
JG et al.).
En algunas de les celdas de la tabla que
hemos construido hemos escrito solo la palabra presente. Esto es debido
a que no hemos encontrado la localización exacta de algunos de los dominios
pero podemos confirmar su existencia gracias a la bibliografía utilizada.
Una vez escogidas las secuencias
mediante Blast y otros artículos científicos ya citados en la bibliografía,
hemos realizado el alineamiento múltiple con ClustalW y el resultado se ha utilizado
para el análisis filogenético con el paquete de programas Phylip: protdist, neighbor-joining,
seqboot y consense. Primero hemos construido un árbol con protdist
y neighbor-joining para conocer las distancias evolutivas entre las
diferentes proteínas de estos organismos. En un segundo paso hemos obtenido el
árbol consenso a partir de las mil matrices de distancias que provienen de los
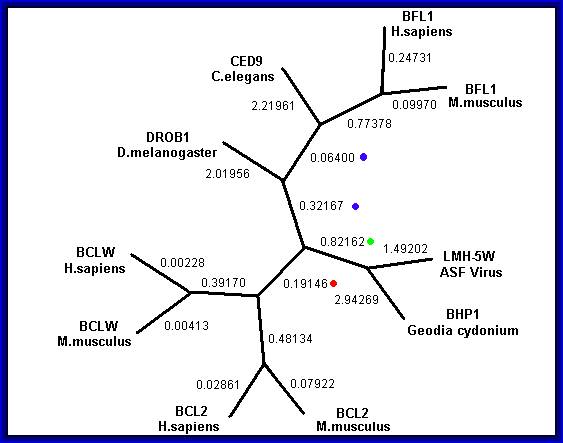
mil alineamientos generados con seqboot. En la imagen vemos el árbol
consenso donde constan las distancias filogenéticas obtenidas con el primer
paso.
La agrupación de los miembros de
Bcl2 y Bclw es la esperada porque los dos pertenecen a la subfamilia
anti-apoptótica de vertebrados y conservan una alta homología. A priori,
creíamos que los Bfl1 se dispondrían juntamente con los miembros
anti-apoptóticos ya que pertenecen a la misma familia funcional, pero el hecho
que no sea así puede explicarse porque solo comparten los dominios BH1 y BH2.
También observamos que BHP1 (G.
cydonium) y LMH-5W (ASFV) se agrupan, aunque están a una distancia
evolutiva considerable porque las dos sólo contienen los dominios BH1 y BH2.
Sobre les proteínas Ced9 y Drob1
no podemos explicar su disposición
razonadamente: Ced9 contiene BH1, BH2 y BH4 y por tanto es una proteína
anti-apoptótica y Drob1 sabemos que es pro-apoptótica y podemos asegurar que
contendrá el dominio BH3 y posiblemente algún otro, aunque no hemos encontrado
ningún tipo de descripción de dominios de esta proteína. Hay la posibilidad que
la localización de éstas en el árbol sea principalmente causado por el efecto
del azar. Este hecho lo explicamos en el siguiente punto.
El valor de bootstrap de
las ramas de un árbol consense está relacionado con la robustez de éstas,
es decir, con el grado en que el azar
interviene en su disposición. En nuestro árbol, la parte que ya se ha comentado
supera un valor de 910 (de 1000 árboles, 910 contienen aquella rama en la misma
posición) por tanto podemos asegurar que el resultado es correcto y no hemos
creído necesario especificarlo en el gráfico. El hecho de analizar organismos
muy alejados filogenéticamente, conlleva a que algunas ramas tengan valores de
robustez más bajos:
![]() ·;
rama que une Bfl1 y Ced9 con el
resto y la que une estas dos más Drob1
con el resto: tienen valores de 300 aproximadamente. Esto es debido a que la
distancia entre C.elegans y D.melanogaster respecto al linaje
vertebrado es considerable; aunque el conjunto de proteínas conserva una función
homóloga, sus secuencias nucleotídicas pueden haberse diversificado mucho. Este
hecho ha provocado un alineamento
condicionado por el azar y unos valores de bootstrap bajos.
·;
rama que une Bfl1 y Ced9 con el
resto y la que une estas dos más Drob1
con el resto: tienen valores de 300 aproximadamente. Esto es debido a que la
distancia entre C.elegans y D.melanogaster respecto al linaje
vertebrado es considerable; aunque el conjunto de proteínas conserva una función
homóloga, sus secuencias nucleotídicas pueden haberse diversificado mucho. Este
hecho ha provocado un alineamento
condicionado por el azar y unos valores de bootstrap bajos.
![]() ·; rama que une los Bcl2 (d’H.sapiens i M.musculus)
y los BclW (d’H.sapiens i M.musculus) con el resto de secuencias:
también tiene un valor de 441. Podría explicarse porque estas cuatro secuencias
son las más homólogas de todo el grupo y un intento de alinearlas con
secuencias más lejanas dificulta que queden relacionadas.
·; rama que une los Bcl2 (d’H.sapiens i M.musculus)
y los BclW (d’H.sapiens i M.musculus) con el resto de secuencias:
también tiene un valor de 441. Podría explicarse porque estas cuatro secuencias
son las más homólogas de todo el grupo y un intento de alinearlas con
secuencias más lejanas dificulta que queden relacionadas.
![]() ·; rama que relaciona las secuencias LMH-5W (African Swine Fever Virus) i
BHP1 (G.cydonium) con el resto: valor muy cercano a 600. En este caso,
pensamos que el motivo vuelve a ser la existencia de una gran distancia
filogenética.
·; rama que relaciona las secuencias LMH-5W (African Swine Fever Virus) i
BHP1 (G.cydonium) con el resto: valor muy cercano a 600. En este caso,
pensamos que el motivo vuelve a ser la existencia de una gran distancia
filogenética.
El hecho que las ramas que
conectan los organismos más alejados filogenéticamente no sean muy robustas nos
obliga a realizar más análisis para comprobar que realmente están relacionadas
evolutivamente con esta familia y, si es así, determinar de qué miembro son
homólogas.
Hemos buscado las secuencias
proteicas en Swissprot de los homólogos de Bcl2 de Epstein-Barr
virus (P03182), African Swine Fever virus (Q07818), Drosophila melanogaster (Q9V9C8) y Caenorhabditis elegans (P41958). Con cada una de estas secuencias hemos
realizado un PSIBlast y de entre los resultados obtenidos se han
buscado en qué proteínas homólogas de vertebrados se observan los scores
más altos.
![]() En el caso del virus africano de la peste
porcina se observa una mayor homología con las proteínas Bclx de vertebrados,
como por ejemplo el Bclx de Xenopus laevis con el mayor score
(50) y el mínimo E-value (1e-05). Con valores muy parecidos encontramos
los Bclx de Bos taurus, Sus scrofa y Mus musculus.
En el caso del virus africano de la peste
porcina se observa una mayor homología con las proteínas Bclx de vertebrados,
como por ejemplo el Bclx de Xenopus laevis con el mayor score
(50) y el mínimo E-value (1e-05). Con valores muy parecidos encontramos
los Bclx de Bos taurus, Sus scrofa y Mus musculus.
![]() Por otra parte, los resultados obtenidos con Epstein-Barr
virus no son concluyentes ya que los homólogos que encontramos de la
familia Bcl2 en vertebrados tienen un E-value próximo a uno.
Por otra parte, los resultados obtenidos con Epstein-Barr
virus no son concluyentes ya que los homólogos que encontramos de la
familia Bcl2 en vertebrados tienen un E-value próximo a uno.
![]() En el caso de C. Elegans este programa
encuentra una alta homología (24%) entre la proteína Ced9 de este organismo y
Bclw de vertebrados. Por ejemplo, con el valor más alto de score (53) y
un E-value más bajo (3e-06) tenemos el Bclw de Rattus
norvegicus, y lo siguen los Bclw de Mus musculus y Homo sapiens.
En el caso de C. Elegans este programa
encuentra una alta homología (24%) entre la proteína Ced9 de este organismo y
Bclw de vertebrados. Por ejemplo, con el valor más alto de score (53) y
un E-value más bajo (3e-06) tenemos el Bclw de Rattus
norvegicus, y lo siguen los Bclw de Mus musculus y Homo sapiens.
![]() El último caso estudiado ha sido el de Drosophila
melanogaster. Encontramos que contiene una homología de un 34% con BOK (de
la subfamilia proapoptótica Bax) de Gallus gallus, con un score de
103 y un E-value 2e-21.
El último caso estudiado ha sido el de Drosophila
melanogaster. Encontramos que contiene una homología de un 34% con BOK (de
la subfamilia proapoptótica Bax) de Gallus gallus, con un score de
103 y un E-value 2e-21.
Como podemos observar los virus
tienen homólogos en vertebrados anti-apoptóticos, el sentido evolutivo
radicaría en el beneficio que obtienen al evitar la muerte celular de sus
huéspedes. En cambio, en D.melanogaster hemos encontrado que la proteína
Drob1 es en realidad un promotor de la apoptosis, esto podría explicar que la
conexión de esta proteína al resto fuera dudosa y, por tanto, entenderíamos
porqué el valor de bootstrap de esta rama es tan bajo.
![]()
![]()